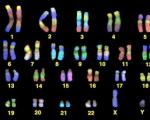
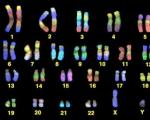
Расшифровка генома человека оказалась бессмысленной. Расшифровка генома человека оказалась бессмысленной Сопоставление данных общего и частного проектов
Спустя десять лет после того, как президент Билл Клинтон объявил об успешном завершении работ над черновым вариантом секвенирования (расшифровки) генома человека, медики говорят, что их ожидания пока не оправдались.
Биологам секвенирование генома преподносило один сюрприз за другим. Но основная цель обошедшегося в $3 млрд проекта «Геном человека», то есть выявление генетических корней таких распространенных заболеваний, как рак и болезнь Альцгеймера, а также создание соответствующих лекарств, по-прежнему остается не достигнутой. Можно сказать, что в результате десятилетних исследований генетики вернулись к исходной точке своих поисков.
Одним из свидетельств ограниченности медицинского применения геномной информации стала недавняя проверка точности предсказания сердечных заболеваний на основании генетических данных. Команда медиков под руководством сотрудницы бостонского Бригемского госпиталя Нины Пэйнтер зафиксировали 101 генетическую мутацию, относительно которых в различных исследованиях по сканированию генома была показана статистическая зависимость с возникновением заболеваний сердца. Но наблюдение 19 000 пациенток в течение 12 лет показало, что эти мутации нисколько не помогают предсказывать возникновение и развитие заболевания. Старомодный метод исследования семейной истории оказался более результативен.
26 июня 2000 года, сообщая миру о завершении работы над черновым вариантом секвенирования генома человека, президент Клинтон сказал, что это достижение означает «революцию в диагностике, профилактике и лечении большинства, если не вообще всех заболеваний человека».
На пресс-конференции тогдашний директор генетического агентства при Национальном институте здоровья Френсис Коллинз пообещал, что в течение 10 лет будет разработана генетическая диагностика заболеваний, а еще через 5 лет появятся новые лекарства. «В длительной перспективе, возможно, через 15-20 лет, - добавил он, - мы станем свидетелями полной революции в медицине».
Фармацевтическая промышленность вложила миллиарды долларов в разработку методов применения раскрытых геномных тайн, и сейчас к выходу на рынок готовятся несколько новых препаратов, созданных с учетом геномной информации. Однако по мере того как фармацевтические компании продолжают вкладывать в исследования генома огромные средства, становится понятным, что генетическая природа большинства заболеваний сложнее, чем предполагалось.
«Геномика имеет огромное значение для науки, но не для медицины», - сказал президент нью-йоркского Мемориального центра раковых исследований Слоун-Кеттеринга, Хэролд Вермус, которому в июле этого года предстоит занять пост директора Национального института раковых исследований.
Последнее десятилетие ознаменовалось потоком открытий патогенных мутаций в геноме человека. Но в отношении большинства заболеваний применение этих открытий позволяет объяснить лишь небольшую часть случаев возникновения патологии.
Целью начатого в 1989 году проекта «Геном человека» было секвенирование, или расшифровка, всех трех миллиардов пар химических оснований, из которых состоит набор инструкций, записанных в человеческом геноме, открытие генетических корней заболеваний и создание на этой основе новых лекарств. После того как секвенирование было закончено, следующим этапом должна была стать идентификация генетических мутаций, увеличивающих риск таких распространенных заболеваний, как рак и диабет.
В то время секвенирование всего генома каждого пациента представлялось слишком дорогим, поэтому Национальный институт здоровья с энтузиазмом воспринял идею, обещавшую более короткий путь к цели: секвенирование только тех мест генома, в которых у многих людей обнаруживаются вариабельные участки ДНК.
За этой идеей стояло теоретическое предположение, что одни и те же распространенные заболевания должны быть следствием также одинаковых и распространенных мутаций. Естественный отбор отсеивает мутации, вызывающие детские патологии, гласила теория, но бессилен против мутаций, возникающих позже в процессе жизни, поэтому последние становятся распространенными. В 2002 году Национальный институт здоровья запустил проект стоимостью $138 млн под названием HapMap, в рамках которого предполагалось составить каталог геномных мутаций, наиболее распространенных среди европейцев, африканцев и жителей Дальнего Востока.
Имея такой каталог, можно выявить мутации, чаще встречающиеся у людей, страдающих определенным заболеванием. В результате были выявлены статистические зависимости между сотнями распространенных генетических мутаций и различными заболеваниями. Но оказалось, что в отношении большинства заболеваний распространенные мутации позволяют объяснить лишь малую часть генетических рисков.
Эрик Лендер, директор Института Брода в Кембридже, штат Массачусетс, и руководитель проекта HapMap, рассказал, что на сегодняшний день установлена связь между 850 участками генома, большинство из которых представляют из себя почти целые гены, и многими распространенными заболеваниями. «Поэтому я убежден, что гипотеза была верной», - говорит он.
Сравнение полных геномов - важнейший метод изучения эволюции, ставший доступным биологам с 1995 года. К настоящему времени полностью прочитано 212 геномов многоклеточных животных. Это уже позволило сделать ряд общих выводов - например, частично реконструировать минимальный геном последнего общего предка животных, а также оценить характерную для современных геномов долю новых генов, свойственных только отдельным систематическим группам.
«Сравнение полных геномов представляет собой единственный удовлетворяющий исследователя путь к реконструкции эволюции», - пишет в книге «Логика случая» знаменитый биоинформатик Евгений Кунин (Eugene Koonin). С категоричностью этого утверждения можно не согласиться, но речь идет и правда о важнейшей вещи. Сравнение полных геномов - метод, который дал биологам совершенно новые возможности для наблюдения эволюционного процесса.
Эпоха сравнения полных геномов началась в 1995 году, когда лаборатория Крейга Вентера (John Craig Venter) опубликовала результат прочтения генома бактерии гемофильного гриппа Haemophilus influenzae (R. D. Fleischmann et al., 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae ). В течение года с того момента было прочитано еще несколько геномов бактерий и один эукариотный - геном дрожжей Saccharomyces cerevisiae. Их можно и нужно было сравнивать, разрабатывая для этого приемы, фиксируя открывающиеся закономерности. За считанные годы родилась новая наука - сравнительная геномика, подобно тому, как в XVIII веке сложилась сравнительная анатомия.
Несколько недель назад американские биологи Кейси Данн (Casey Dunn) и Джозеф Райан (Joseph F. Ryan) выпустили очень краткий - на пяти страничках - обзор данных , полученных из прочитанных на данный момент полных геномов многоклеточных животных. Их уже столько, что можно сделать первые выводы, касающиеся генетической эволюции животных в целом.
Данн и Райан определили предмет своего интереса совершенно четко: макроэволюция ядерных геномов. Оговорка о макроэволюции означает, что нас сейчас интересуют геномные различия между крупными группами организмов, а не те, что можно найти между близкими видами, и тем более не внутривидовые. Оговорка про ядерные геномы означает, что мы не рассматриваем, например, митохондриальную ДНК (которую тоже очень интересно изучать, но это особая тема). Итак, что нам известно о ядерных геномах животных?
Сначала сухие цифры. На момент публикации статьи Данна и Райана число полностью прочитанных геномов животных составляло 212 (даже за прошедшее с тех пор короткое время их могло стать уже больше). Подразумевается, что каждый геном принадлежит отдельному виду. Двести двенадцать видов - это меньше двух сотых долей процента от общего числа видов современных животных, которое составляет примерно 1,5 миллиона. Кроме того, разные группы животных представлены в этой выборке очень неравномерно. Во-первых, 83% прочитанных геномов принадлежат позвоночным. Во-вторых, наблюдается явный перекос в сторону животных с маленькими геномами, работать с которыми технически гораздо проще. Напомним, что размеры геномов разных животных могут отличаться на три порядка, то есть в тысячи раз (см. Геномы хвостатых амфибий с самого начала были большими , «Элементы», 24.06.2015). Обладателям гигантских геномов, например, двоякодышащим рыбам, придется еще какое-то время подождать своей очереди.
Тем не менее, все крупные эволюционные ветви животных современной выборкой охвачены, так что ее можно считать вполне представительной (рис. 1).
Уточним это утверждение. На эволюционном древе есть семь крупных ветвей, без упоминания которых не обходится ни один разговор о животном царстве (если, конечно, его хотят охватить в целом). Это губки , гребневики , пластинчатые , стрекающие , линяющие , спиральнодробящиеся и вторичноротые . Так вот, во всех этих ветвях без исключения есть представители, полные геномы которых уже прочитаны и внесены в электронные базы. В большинстве ветвей - и не по одному.
Все геномы животных гораздо более похожи друг на друга, чем можно было бы ожидать, исходя из внешних различий их обладателей. Например, размер генома очень слабо коррелирует со сложностью организма. Конечно, упоминая в биологии «сложность», приходится всегда оговаривать, что понятие это абстрактное и трудно определимое. Но при любом его понимании не приходится сомневаться, например, в том, что человек, одна только нервная система которого насчитывает 86 миллиардов нейронов, сложнее круглого червя Caenorhabditis elegans , у которого нейронов 302 штуки. В догеномную эпоху высказывались предположения, что у человека примерно 140 тысяч генов, но сейчас ясно, что их всего 20–25 тысяч (см. А. Панчин, 2015. Сколько мусора в нашей ДНК?). А число генов ценорабдитиса - 19 735 (L. D. W. Hillier et al., 2005. Genomics in C. elegans : So many genes, such a little worm). По числу генов человек и ценорабдитис мало отличаются друг от друга.
Любой геном можно охарактеризовать двумя главными параметрами: размер генома (C) и число генов (G). Размер генома измеряется в парах нуклеотидов, число генов - просто в штуках. Широко известно, что размер генома может очень сильно - на порядки - отличаться у организмов, явно близких по сложности устройства тела (например, у хвостатых и бесхвостых земноводных). Это явление называют «парадоксом значений C».
А вот с числом генов все наоборот. Оно часто бывает примерно одинаковым (во всяком случае, сравнимым) у организмов, отличающихся чуть ли не по всем признакам устройства тела, какие только можно придумать. Этот факт получил название «парадокса значений G» (M. W. Hahn, G. A. Wray, 2002. The g-value paradox). Теоретически объяснять его можно по-разному, но в любом случае это один из главных выводов, сделанных с помощью чтения полных геномов. Другими способами надежно узнать число генов нельзя. Например, если мы измеряем химическими методами количество ДНК в клетке, то сразу получаем информацию о размере генома, но отличить гены от некодирующих последовательностей (которых в геномах эукариот всегда много) таким образом невозможно.
Еще одна задача, которую нельзя решить без исследований полных геномов - это реконструкция генома общего предка всех многоклеточных животных. Сравнение геномов разных животных, а также их одноклеточных родственников, позволяет в принципе определить минимальный набор генов, которыми этот предок должен был обладать. На данный момент можно сказать, что у последнего общего предка всех животных было по меньшей мере 6289 генных семейств, общих у него с некоторыми одноклеточными, и 2141 генное семейство, уникальное для животных (напомним, что генное семейство - это группа генов, происходящих от одного гена-предшественника путем его удвоений; см., например: Предки губок могут оказаться сложнее, чем предполагалось , «Элементы», 27.10.2015). Получается, что у общего предка животных было минимум 8430 генов. Правда, это - в случае, если каждое генное семейство состояло только из одного гена, что маловероятно. На самом деле генов, скорее всего, было больше, но все же эти цифры дают какую-никакую точку отсчета. И в любом случае, они не окончательные - ведь анализ геномов продолжается.
Некоторые выводы касаются отдельных групп генов и белков. Например, белки клеточной адгезии (cell adhesion molecules, CAM), обеспечивающие сцепление и взаимодействие клеток друг с другом, оказались вовсе не уникальными для животных - они появились у наших одноклеточных родственников, которым были нужны, вероятно, ввиду их сложного жизненного цикла (см. H. Suga et al., 2013. The Capsaspora genome reveals a complex unicellular prehistory of animals). А вот многие гены и белки, обеспечивающие внутриклеточную сигнализацию, для животных как раз уникальны.
От генома последнего общего предка многоклеточных животных, естественно, произошли - путем многократных копирований с изменениями - геномы всех животных, доживших до современности. Так и сформировалось ветвистое эволюционное древо, которые мы теперь видим. События, меняющие геномы в ходе эволюции, очень разнообразны: дупликации разных типов, перестройки устойчивого совместного расположения генов (синтении), умножение или потеря генов тех или иных семейств, всевозможные вставки, модификации регуляторных областей ДНК, перенос мобильных генетических элементов , изменение состава повторяющихся последовательностей, таких как сателлитная ДНК , и многое другое. Все эти события оставляют следы, которые наслаиваются друг на друга. Их можно найти и прочитать, примерно так же, как можно найти и прочитать следы правок в обычном тексте. Ведь геном - тоже текст (хотя и не только текст, конечно). Геном любого животного заключает в себе целую летопись изменений, многие из которых можно довольно точно распознать и датировать. Сплошь и рядом это относится даже к изменениям, которые произошли много сотен миллионов лет назад. Текст может сохранить все.
Сейчас широко известно, что генный «инвентарь» многоклеточных животных эволюционно очень консервативен. Сплошь и рядом одни и те же гены используются для близких функций и у человека, и у относительно простых существ наподобие того же ценорабдитиса. Однако это - далеко не вся правда. В любом эукариотном геноме есть 10–20% генов, не похожих ни на какие гены смежных (и любых других) групп организмов (K. Khalturin et al., 2009. More than just orphans: are taxonomically-restricted genes important in evolution?). Такие гены часто называют «генами-сиротами» (orphan genes), имея в виду, что для них не удается найти генов-предков. Но все-таки корректнее называть их таксономически ограниченными генами (taxonomically-restricted genes, TRG). Таксономически ограниченный ген - это действительно новый ген, появившийся в эволюции одной-единственной группы организмов. Например, есть гены, свойственные только позвоночным, только некоторым насекомым или даже только мышам. Естественно, тут возникают интересные вопросы. Во-первых, откуда новые гены берутся? И, во-вторых, какие у них обычно бывают функции?
На вопрос, откуда берутся новые гены, самый распространенный ответ простой: новые гены берутся из старых. Например, в результате случайной дупликации образуются две копии некоего гена, в первой из них мутации накапливаются медленнее, во второй быстрее, в итоге вторая копия постепенно становится новым геном с новой функцией. Это действительно очень частый механизм создания новых генов, но не единственный. По-видимому, новые гены в принципе могут образоваться из чисто регуляторных участков ДНК или даже из некодирующих последовательностей (D. Tautz, T. Domazet-Loso, 2011. The evolutionary origin of orphan genes). Иногда на это накладываются дополнительные события, например вставка в новый ген небольшого куска другого, старого гена. Кстати, установлено, что «молодые» гены обычно кодируют более короткие белки, чем «старые». Получается, что долгоживущий ген как бы накапливает груз, состоящий из случайных вставок - которые, впрочем, в свою очередь могут подвергаться отбору и обретать новые функции. В любом случае, образование новых генов, как и большинство процессов в биологии, не сводится к какому-либо одному механизму. Таких механизмов несколько, и они могут сочетаться.
Что касается функций таксономически ограниченных генов, то они бывают очень разнообразными. Роднит их одно: большинство этих функций - специфические для данной группы животных, не встречающиеся за ее пределами.
Например, свой набор таксономически ограниченных генов есть у типа стрекающих , к которому относятся медузы и полипы (включая гидр и кораллов). Пожалуй, самая яркая особенность стрекающих животных - стрекательные клетки (книдоциты), в честь которых этот тип получил свое название. Книдоциты - одни из самых сложных клеток, какие только можно найти в животном мире. В каждом книдоците находится свернутая стрекательная нить с ядовитым веществом и механизмом выбрасывания. Книдоциты стрекающих очень разнообразны, их насчитывается более 30 типов, а вот ни у каких других животных подобных клеток нет. Так вот, оказалось, что очень многие гены, свойственные только стрекающим, связаны со специфическими органеллами стрекательных клеток. Например, целая группа таких генов кодирует миниколлагены - уникальные короткие белки, входящие в состав стенки внутриклеточной капсулы, в которой размещается стрекательная нить. У одного из видов пресноводной гидры найден 41 ген, активный только в стрекательных клетках и не похожий ни на какие гены других типов животных. Выходит в общем логично: новые гены обеспечивают эволюционные новшества, проявляющиеся уже на уровне организма (рис. 2).
Любой геном - это в некотором смысле архив, документирующий события, происходившие на всем протяжении эволюционной линии от последнего общего предка всех живых организмов (last universal common ancestor, LUCA) до обладателя этого генома. Например, в геноме мыши запечатлены следы событий, происходивших по всей непрерывной цепочке предков и потомков от LUCA до этой самой мыши. Конечно, некоторые следы в ходе эволюции стираются бесследно, и с этим ничего не поделаешь. Но очень многие сохраняются. Причем все эти следы заключены внутри сложной упорядоченной структуры, дающей возможность их послойно датировать. В этом отношении работа сравнительного геномика напоминает стратиграфию или археологию.
При всем величии сравнительной геномики ее никак нельзя назвать «альфой и омегой» современной эволюционной биологии. Данн и Райан совершенно справедливо замечают, что далеко не всех биологов, занятых сравнительным анализом геномов, интересует геномная эволюция сама по себе. Гораздо чаще история геномов служит посредником (proxy) для понимания других аспектов эволюции, связанных с фенотипом и организмом. Подобным же образом историка, методами палеографии и источниковедения изучающего средневековые документы, чаще всего интересуют не тексты как таковые, а социально-исторические процессы, отображение которых в этих текстах можно найти.
Первое, что заметила Дэбби Джорд (Debbie Jorde) у своей новорожденной дочери – это неестественно согнутые руки. Но у нее были и другие дефекты развития: волчья пасть, восемь пальцев на руках, восемь пальцев на ногах и отсутствие нижних век. Ребенку поставили диагноз синдрома Миллера – настолько редкого заболевания, что врачи долгое время считали, что оно не передается из поколения в поколение, а развивается только в результате спонтанных мутаций. У сына Дэбби Джорд, рожденного тремя годами ранее, были те же симптомы. Врачи уверяли, что вероятность рождения второго ребенка с таким же синдромом составляла один на миллион. Но они ошибались.
Муж Дэбби Линн Джорд (Lynn Jorde), генетик из Университета Юты (University of Utah) (Солт-Лейк Сити, США), все еще вспоминает слова врачей, сказавших, что для данного заболевания слишком мало данных, чтобы предсказать риск его развития.
Теперь, благодаря секвенированию нового поколения, Дэбби и ее дети знают свой генетический риск. Дэбби, ее муж и двое уже взрослых детей – Хизер и Логан Мадсен, стали первой семьей, чьи геномы были полностью секвенированы в 2009 г. .
На протяжении 6 месяцев ученые проводили перекрестный анализ огромного количества последовательностей ДНК из 4 геномов. Параллельное секвенирование геномов других пациентов с синдромом Миллера позволило выявить ген, ответственный за развитие данного заболевания. Им оказался ген DHODH , кодирующий белок, участвующий в синтезе нуклеотидов. Выяснилось, что заболевание носит рецессивный характер наследования. В данном случае оба родителя являлись носителями одной мутантной копии гена, следовательно, шансы рождения больного ребенка составляли 25%. Генетический анализ позволил выявить у детей еще одно рецессивное наследственное заболевание – первичную цилиарную дискинезию , влияющую на развитие легких. «До этого мы не могли понять, почему у детей так часто возникает пневмония» , - говорит Дэбби.
Семьи наподобие Джорд составляют небольшую, но все растущую группу людей, в основном с редкими заболеваниями, чьи геномы были секвенированы с целью постановки диагноза и изучения конкретной болезни. Хотя знание о полной последовательности ДНК никак не повлияло на лечение Хизер и Логана, многие люди секвенируют свой геном именно с этой целью. В прошлом году мальчику из Висконсина (США) на основании результатов секвенирования был имплантирован жизненно необходимый трансплантат красного костного мозга . Женщине с лейкемией, а также близнецам с редким наследственным заболеванием, результаты секвенирования помогли назначить адекватное лечение (см. ).
До настоящего времени секвенировать свой геном могли лишь люди, лично знавшие ученых, заинтересованных в клинической генетике, либо семьи с очень редкими заболеваниями, наподобие Джорд. Но теперь, когда полногеномное секвенирование становится все дешевле и доступнее, по всему миру запускаются клинические программы для анализа секвенированных последовательностей. Компания Illumina (Сан-Диего, США), продающая секвенаторы и программы для их обработки, предлагает услугу полногеномного секвенирования всего за 7500 долларов США для людей с тяжелыми наследственными заболеваниями, и за 10000 долларов США для онкологических больных, которым необходимо секвенировать геномы раковых и здоровых клеток.
Поскольку цены на секвенирование продолжают падать, через некоторое время определение последовательности целого генома и ее анализ будет сродни процедуре магнитно-резонансной томографии (МРТ). «Это будет так же просто, как и проведение любого другого медицинского анализа , - говорит Дэвид Бик (David Bick), клинический генетик из Медицинского Колледжа Висконсина (Medical College of Wisconsin , США), - Однако, в отличие от результатов большинства медицинских тестов, секвенирование генома предоставит огромное количество трудно интерпретируемых данных, из которых далеко не вся информация будет необходима для диагностики или лечения заболеваний пациента. Кроме того, пациент получит нежелательную информацию о предрасположенности к некоторым другим заболеваниям» .
«Анализ геномов и консультирование пациентов и их семей может стать слишком тяжелым бременем для клиницистов. То, что было опробовано на нескольких пациентах, нельзя сразу перенести на широкое клиническое использование , - говорит Эрик Грин (Eric Green), директор Национального Исследовательского Института Генома Человека (National Human Genome Research Institute, NHGRI) в Бетесде (США), - Хотя некоторые отдельные случаи очень показательны» .
Николас Волькер (Nicholas Volker) родился с недиагностируемым заболеванием кишечника, проявляющимся периодическим образованием в нем фистул, что требовало постоянного хирургического вмешательства. К тому времени, как Волькеру исполнилось 3 года, он перенес более 100 операций. Врачи предполагали, что у ребенка иммунный дефицит, и трансплантат красного костного мозга будет правильным решением проблемы. Но большое количество анализов, включая секвенирование нескольких генов, не подтвердило поставленный диагноз. После многочисленных обсуждений группа ученых из Медицинского Колледжа Висконсина решила секвенировать его экзом – последовательности, непосредственно кодирующие молекулы РНК и составляющие 1-2% генома.
Исследовав последовательности с помощью специальных программ, ученые выявили мутацию на Х-хромосоме в гене XIAP (X-linked inhibitor of Apoptosis). Известно, что дефицит белка, кодируемого данным геном, определяет высокий риск развития смертельного иммунного заболевания, и трансплантация красного костного мозга в данном случае просто необходима.
Первая программа по сравнительному полногеномному секвенированию была создана в Медицинском Колледже Висконсина. В настоящее время клинические генетики колледжа концентрируют свое внимание на пациентах с редкими наследственными заболеваниями, для которых идентификация генетических дефектов позволяет определить курс лечения. Для некоторых пациентов, принявших участие в программе, расходы согласились оплатить страховые компании. «Их обоснование довольно просто , - говорит Тина Хэмбак (Tina Hambuch), главный разработчик из клинической лаборатории компании Illumina , - Полногеномное секвенирование может обойтись дешевле серии генетических тестов, а также прояснить, потребуется ли дорогостоящее лечение» .
Примеру Медицинского Колледжа Висконсина последовали и другие институты. В Великобритании Центр Генетики Человека Wellcome Trust Centre for Human Genetics (Оксфорд) планирует отсеквенировать геномы 500 человек. Программа Недиагностируемых Заболеваний (The Undiagnosed Diseases Program) Национальных Институтов Здоровья в Бетесде (США) осуществляет секвенирование пациентов с 2008 г. Было проанализировано более 140 экзомов и 5 геномов с целью поиска молекулярных основ трудно диагностируемых заболеваний.
Генетические основы многих заболеваний все еще не изучены. Он-лайн ресурс OMIM (Online Mendelian Inheritance in Man) содержит характеристики более 7000 заболеваний человека, и только для половины из них описаны молекулярные механизмы. По словам Эрика Грина, в ближайшее время Национальный Исследовательский Институт Генома Человека выдаст грант на финансирование нескольких центров секвенирования, занимающихся исследованием молекулярных причин заболеваний.
По словам ученых, результаты секвенирования проще будет использовать для диагностики и терапии рака, нежели редких наследственных заболеваний. Клиницисты уже проводят детальный анализ некоторых раковых опухолей, чтобы адаптировать терапию под генетические характеристики пациента. Например, в геноме конкретной раковой клетки иногда можно обнаружить дефекты химических сигнальных путей, исходя из чего можно подобрать определенный препарат. Для стандартных методов диагностики это не представляется возможным.
В 2007 г. 78-летний канадец с раком языка, давшим метастазы практически по всему организму, проходил лечение в онкологическом центре British Columbia Cancer Agency (Ванкувер, Канада). Для этого типа рака не существовало одобренного метода лечения, и врачи убедили ученых онкологического центра провести секвенирование генома опухолевых клеток. Ученые также проанализировали транскриптом, что позволило изучить не только последовательность ДНК, но и количество продуцируемой опухолевыми клетками РНК. Сравнив полученные данные с данными по другим раковым опухолям и последовательностью генома нормальных клеток, ученые сконцентрировали свое внимание на гене RET , который был дуплицирован в геноме раковых клеток и транскрибировался в большое количество молекул мРНК. Известно несколько препаратов, ингибирующих белок, кодируемый данным геном. По словам Марко Марра (Marco Marra), директора онкологического центра, после долгих размышлений предпочтение было отдано препарату сунитинибу. На несколько месяцев развитие рака удалось приостановить, после чего опухоль снова начала метастазировать. Анализ вновь образовавшихся опухолей показал, что в злокачественных клетках активировались сигнальные пути, вызывающие рак, что сделано опухоль резистентной к первому препарату, но, возможно, чувствительной к другим. Но, к сожалению, было слишком поздно: пациент умер.
Научная группа Марра в настоящее время работает над созданием проекта по улучшению диагностики другого типа рака – острой миелоидной лейкемии – с помощью секвенирования генома и транскриптома. Вдохновленные примером Марра, генетики из Университета Вашингтона (Washington University) в Сент-Луисе (США) также осуществляют секвенирование геномов онкологических больных с целью подбора эффективной противораковой терапии.
Однако перевод полногеномного секвенирования из области исследований в клиническую практику сталкивается с определенными препятствиями. В отличие от исследовательской деятельности, секвенирование ДНК, предназначенное для постановки диагноза, должно проводиться в аккредитованных лабораториях, таких как в компании Illumina . Наблюдательные комитеты, осуществляющие надзор за исследованиями на человеке, еще не достигли консенсуса о том, требуется ли одобрение процедуры клинического секвенирования, а Управлению по Контролю за Качеством Пищевых Продуктов и Лекарственных Средств США (US Food and Drug Administration , FDA) еще предстоит решить, как регулировать приближающуюся волну клинического секвенирования.
Многие ученые и клиницисты опасаются, что в системе здравоохранения не достаточно людей, хорошо разбирающихся в области геномики и биоинформатики, чтобы адекватно интерпретировать огромный поток информации. По словам экспертов, данные о заболеваниях человека разбросаны по многочисленным научным статьям и базам данных, в которых порою трудно разобраться. Кроме того, анализ последовательности представляет собой весьма трудоемкую работу. По словам Хэмбак, в тех нескольких научно-исследовательских проектах, в которых принимала участие компания Illumina , только выявление всех полиморфных вариантов генома заняло 2-3 недели. «Это трудоемкая работа для высокопрофильных специалистов» , - комментирует Хэмбак.
Некоторая информация может оказаться лишней для пациентов. Медицинские генетики и специалисты по этике уже давно обеспокоены поиском генетических маркеров, связанных с риском заболеваний, не поддающихся лечению. Имея полную последовательность генома, вероятность получения такой «лишней» информации взмывает вверх. Ситуация также сложна для юных пациентов. Имеют ли родители право решать за детей, какую информацию им стоит знать, а какую – нет.
По этим причинам Стефан Кингсмор (Stephen Kingsmore), работающий в клинике Children"s Mercy Hospital в Канзас-Сити (США), утверждает, что клиническое секвенирование должно носить ограниченный характер. В настоящее время его научная группа разрабатывает метод одновременной детекции панели мутаций, связанных с более 600 рецессивными заболеваниями.
Но некоторые генетики считают, что «поезд не остановить». «После того как была продемонстрирована информативность данной технологии, я думаю, она будет широко использоваться» , - говорит Хакон Хаконарсон (Hakon Hakonarson), в настоящее время запускающий программу клинического секвенирования в Детском Госпитале Филадельфии (Children"s Hospital of Philadelphia , США).
Семья Джорд все еще размышляет над тем, что им дало секвенирование. И хотя его результаты все равно не повлияли на терапию, если бы Дэбби и Линн знали о проблемах с легкими детей раньше, можно было бы избежать опасной операции, которую пришлось перенести Хизер и Логану, чтобы снизить вероятность рецидива пневмонии.
По мнению Линна, клиническое секвенирование ждет успех. «Но я прирожденный оптимист» , - добавляет он.
Оригинальный текст: Brendan Maher
История становления и развития психогенетики как научной дисциплины.
Древние врачи и натурфилософы строили гипотезы по поводу наследственности по видимым данным: внешние сходства детей и родителей, характер, голос, походка, а также болезни и уродства.
Всю историю становления и развития психогенетики можно условно поделить на пять этапов (1973 г. В.Томпсон и Г.Уайльд).
Первый этап (1865 - начало 1900-х) – Гальтон и его последователи. В 1865 первая научная публикация по психогенетике "Наследственный талант и характер" – идеи наследуемости психических особенностей, одаренности и. Вслед за этим вышла его знаменитая книга "Наследственный гений" (1869), а также статьи "Люди науки, их воспитание и характер" (1874) и "История близнецов как критерий относительной силы природы и воспитания
Второй этап - до конца 30-х гг. ХХ столетия - характеризуется интенсивным развитием методологии психогенетики. Были разработаны надежные методы определения зиготности близнецов (Siemens H., 1927). В 20-е гг. в методический арсенал психогенетики прочно вошел метод приемных детей, который и сейчас, наряду с близнецовым, является одним из основных.
Благодаря совместным усилиям генетиков и математиков, совершенствовались методы количественной генетики. большинство психологических признаков относятся к категории количественных, т.е. требуют измерения и применения вариационно-статистических методов.
На третьем этапе (до конца 60-х гг.) психогенетика развивалась экстенсивно. Это был период накопления фактического материала. Продолжала развиваться генетика поведения животных. В 1960 г. вышла первая обобщающая монография по генетике поведения.В этом же году была основана "Ассоциация генетики поведения" , начал выходить журнал "Генетика поведения" .Это означало, что генетика поведения окончательно оформилась как самостоятельная область науки.
Четвертый этап (до конца 80-х гг.) вновь характеризуется смещением акцентов на развитие методологии психогенетики. совершенствование компьютерных технологий. В этот период начали интенсивно развиваться новые генетико-математические методы (структурное моделирование, метод путей). Психогенетика получила для своих исследований мощный инструмент, который позволял в короткие сроки проводить обработку значительных массивов данных и проверять самые сложные гипотезы.
Неослабевающий долгие годы интерес к исследованию наследуемости интеллекта уступает место другим характеристикам человеческой индивидуальности (когнитивным стилям, темпераменту, личности, психофизиологическим особенностям, различным нарушениям развития). Все более тщательно изучаются различные аспекты средовых влияний, создаются специальные методики для изучения семейной среды. Во всем мире начинают закладываться лонгитюдные проекты исследования близнецов и приемных детей, позволяющие проследить траектории развития и генетическую преемственность.
Пятый этап - современный - охватывает 90-е гг. ХХ в. и начало нынешнего, т.е. по времени совпадает с интенсивной работой над проектом "Геном человека". Преобладающим направлением сейчас можно считать геномное. Р. Пломин -в генетике поведения более перспективным является движение "от поведения к генам", включая взаимодействия и корреляции между генотипом и средой, а также возможности коррекции генетических нарушений с помощью средовых воздействий, т.е. "средовой инженерии".
Проект «Геном человека» цели, задачи и достижения
Проект по расшифровке генома человека - международный научно-исследовательский проект, главной целью которого было определить последовательность нуклеотидов, которые составляют ДНК и идентифицировать 20-25 тыс. генов в человеческом геноме.
Проект начался в 1990 году, под руководством Джеймса Уотсона. Целью проекта по расшифровке генома человека является понимание строения генома человеческого вида.
Изначально планировалось определение последовательности более трёх миллиардов нуклеотидов, содержащихся в гаплоидном человеческом геноме. Затем несколько групп объявили о попытке расширить задачу до секвенирования биополимеров - определение их аминокислотной или нуклеотидной последовательности. В результате секвенирования получают формальное описание первичной структуры линейной макромолекулы в виде последовательности мономеров в текстовом виде диплоидного генома человека .
Геном любого отдельно взятого организма (исключая однояйцевых близнецов) уникален, поэтому определение последовательности человеческого генома в принципе должно включать в себя и секвенирование многочисленных вариаций каждого гена. Однако, в задачи проекта «Геном человека» не входило определение последовательности всей ДНК, находящейся в человеческих клетках; а некоторые гетерохроматиновые области (в общей сложности около 8 %) остаются несеквенированными до сих пор.
Создание детальной физической карты генома человека;
Создание физических карт всех хромосом человека и хромосом ряда модельных организмов;
Определение полной последовательности ДНК человека и ряда модельных организмов;
Развитие методологии и инфраструктуры для хранения, анализа и распределения полученной информации;
Создание технологии, необходимой для достижения перечисленных целей.
Геном был разбит на небольшие участки, примерно по 150 000 пар нуклеотидов в длину. Эти куски затем встраивали в вектор, известный как Искусственная бактериальная хромосома или BAC. Эти векторы созданы из бактериальных хромосом, измененных методами генной инженерии. Векторы, содержащие гены, затем можно вставлять в бактерии, где они копируются бактериальными механизмами репликации. Каждый из кусочков генома потом секвенировали раздельно методом дробовика, и затем все полученные последовательности собирали воедино уже в виде компьютерного текста. Размеры полученных больших кусков ДНК, собираемых для воссоздания структуры целой хромосомы, составляли около 150 000 пар нуклеотидов. Такая система известна под именем «иерархического метода дробовика», потому что вначале геном разбивается на куски разного размера, положение которых в хромосоме должно быть заранее известно.
Сущ. многочисленные определения «полной последовательности человеческого генома». Согласно некоторым из них, геном уже полностью секвенирован, а согласно другим, этого ещё предстоит добиться. остаётся несколько регионов, которые считаются незаконченными:
Прежде всего, центральные регионы каждой хромосомы, известные как центромеры, которые содержат большое количество повторяющихся последовательностей ДНК
Во-вторых, концы хромосом, называемые теломерами, также состоящие из повторяющихся последовательностей, и по этой причине в большинстве из 46 хромосом их расшифровка не завершена. Точно не известно, какая часть последовательности остаётся не расшифрованной до теломер, но как и с центромерами, существующие технологические ограничения препятствуют их секвенированию.
В-третьих, в геноме каждого индивидуума есть несколько локусов, которые содержат членов мультигенных семейств, которые также сложно расшифровать с помощью основного на сегодняшний день метода фрагментирования ДНК.
Бо́льшая часть остающейся ДНК сильно повторяющаяся, и маловероятно, что она содержит гены, однако это останется неизвестным, пока они не будут полностью секвенированы. Понимание функций всех генов и их регуляции остается далеко неполным.
Все люди имеют в той или иной степени уникальные геномные последовательности. Поэтому данные, опубликованные проектом «Геном человека», не содержат точной последовательности геномов каждого отдельного человека.
Почти все цели, которые ставил перед собой проект, были достигнуты быстрее, чем предполагалось. Проект поставил разумную, достижимую цель секвенирования 95 % ДНК. Исследователи смогли секвенировать 99,99 % человеческой ДНК.