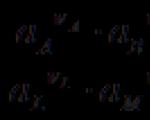Electrochemistry. Galvanic cell Electrochemical cell
Electrochemical elements. Electromotive force. Thermodynamics of a galvanic cell. EMF measurement.
Electric double layer, mechanism of occurrence and structure.
GALVANIC ELEMENTS. EMF.
When an electric current passes through an electrolyte, electrochemical reactions occur on the surface of the electrodes. The occurrence of electrochemical reactions can be generated by an external current source. The opposite phenomenon is also possible: electrochemical reactions occurring on two electrodes immersed in an electrolyte generate an electric current, and the reactions occur only in a closed circuit (when current passes).
Electrochemical (or galvanic) cell is a device for producing electric current through electrochemical reactions. The simplest electrochemical element consists of two metal electrodes (conductors of the first kind), lowered into an electrolyte (conductor of the second kind) and connected to each other by a metal contact. Several electrochemical elements connected in series form electrochemical circuit .
The most important quantitative characteristic of an electrochemical element is electromotive force(EMF, E), which is equal to the potential difference correctly open element (one in which conductors of the first kind from the same material are connected to the final electrodes of the element).
If, when an electric current passes in different directions, the same reaction occurs on the surface of the electrode, but in opposite directions, then such electrodes, as well as the element or circuit made up of them, are called reversible . The emf of reversible elements is their thermodynamic property, i.e. depends only on T, P, the nature of the substances that make up the electrodes and solutions, and the concentration of these solutions. An example of a reversible element is Daniel-Jacobi element :
(-) Cu çZn çZnSO 4 ççCuSO 4 çCu (+)
in which each electrode is reversible. When the element operates, the following reactions occur: Zn ® Zn 2+ + 2 e, Cu 2+ + 2 e® Cu. When a current of infinitesimal strength is passed from an external source, reverse reactions occur on the electrodes.
An example of an irreversible element is Volta element :
(-) Zn ç H 2 SO 4 ç Cu (+)
When the element operates, the following reactions occur: Zn ® Zn 2+ + 2 e, 2H + + 2 e® H 2 . When passing current from an external source, the electrode reactions will be: 2H + + 2 e® H 2 , Cu ® Cu 2+ + 2 e .
The EMF of an electrochemical element is a positive value, because it corresponds to a certain spontaneous process that produces positive work. The reverse process, which cannot occur independently, would correspond to a negative EMF. When composing a chain of electrochemical elements, the process in one of the elements can be directed so that it is accompanied by the expenditure of work from the outside (non-spontaneous process), using for this the work of another element of the chain in which a spontaneous process occurs. The total emf of any circuit is equal to the algebraic sum of positive and negative quantities. Therefore, it is very important when writing a circuit diagram to take into account the signs of the EMF, using the accepted rules.
The emf of the electrochemical circuit is considered positive, if, when writing the circuit, the right electrode is charged positively relative to the left (during the operation of the circuit, cations pass in the solution from the electrode written on the left towards the electrode written on the right, and electrons move in the same direction in the external circuit). Example.
THERMODYNAMICS OF A GALVANIC CELL .
Let the reaction proceed reversibly and isothermally in an electrochemical system:
n A A + n B B + ... ± nF Û n L L + n M M + ... ±
The electrical energy generated by the element is equal to the useful work A¢ of the total process. The useful work A¢ of the reversible process is maximum and at P, T = const is equal to the decrease in the isobaric potential of the system:
DG P, T = nFE P, T
E P , T - reversible EMF of the system.
E P,T = -DG P,T / nF , E V,T = -DF V,T / nF
Thus, by measuring the EMF of the element and its temperature coefficient, it is possible to find the values of DG and DS for the total process occurring in the galvanic cell. This process is spontaneous, hence DG< 0.
Using the Gibbs-Helmholtz equation, we can calculate the change in enthalpy of the process:
DH = DG - T = -nFE P + TnF
nFE P = -DH + nFT = + nFT
nFE V = -DU + nFT = + nFT
From the equations it follows that the relationship between electrical energy reversibly generated or absorbed in an electrochemical system and the thermal effect of the reaction occurring in it depends on the sign and magnitude of the temperature coefficient of the emf dE/ dT :
1. IfdE / dT > 0 , then nFE > (DG > DH) and the system will convert into electrical energy not only the amount of heat that corresponds to the thermal effect of the reaction, but also additional heat - Peletier's warmth Q P = nFT dE/ dT borrowed from the environment. Under adiabatic conditions (under conditions of thermal insulation, when exchange with the environment is impossible), the T of the system decreases. The cooling of the system is especially noticeable if dE/ dT > 0 < 0 (реакция эндотермична).
2. IfdE / dT < 0 , then nFE< (DG < DH) и часть теплоты реакции будет рассеиваться в виде теплоты Пелетье. В адиабатическом режиме система будет нагреваться.
3. IfdE / dT = 0 , then DG = DH and nFE = - the electrical energy produced reversibly by the system is equivalent to the thermal effect of the chemical reaction. This relationship is known as Thomson's principle (rule) .
To calculate the EMF, the equations can be rewritten as:
When using equations, it must be remembered that they valid only for reversible electrochemical systems, therefore, when studying the dependence of EMF on T, it is necessary to avoid the use of electrochemical systems with liquid boundaries, because the diffusion potentials arising on them are not equilibrium.
Let us associate the EMF of the element with the equilibrium constant of the reaction occurring in the element. Chemical reaction isotherm equation:
DG = RT ln K a - RT
E = - = ln K a -
The first term on the right side of the equation for given P, T is a constant value; it can be denoted by E o. E o - standard EMF of an element (electrochemical system) , i.e. EMF at all a i = 0.
E = E o + ln
 = E o + 2.303 lg
= E o + 2.303 lg
Thus, the EMF of an electrochemical system is a function of the activities of participants in the electrochemical reaction. The above equations make it possible to calculate the values of DG and K A based on experimental values of E and, conversely, calculate E, knowing the thermodynamic characteristics of the chemical reaction.
EMF MEASUREMENT .
To measure the equilibrium (reversible) value of the EMF of an electrochemical element, it is necessary that the process occurs infinitely slowly, i.e. so that the element operates at an infinitesimal current. This condition is met in the compensation method, which is based on the fact that the element is connected in series against an external potential difference and the latter is selected so that there is no current in the circuit. Then the external potential difference is equal to the EMF of the circuit.
Using the compensation method, you can directly measure the value of the EMF, but this is a rather complicated operation, therefore in laboratory practice they prefer to compare the EMF of the element being studied with the EMF of the so-called standard (normal) elements, which is carefully measured at different T. This comparative method is also compensation.
The basic normal element is saturated Weston element .
(EMF measurement circuit - independently).
STRUCTURE OF ELECTRODE BOUNDARY - SOLUTION. DOUBLE ELECTRIC LAYER .
When a conductor of the first kind comes into contact with an electrolyte, a electrical double layer . As an example, consider a copper electrode immersed in a CuSO 4 solution. The chemical potential of copper ions in a metal at a given T can be considered constant, while the chemical potential of copper ions in solution depends on the salt concentration; in general, these chemical potentials are not the same.
Let the concentration of CuSO 4 be such that > . Then, when the metal is immersed in the solution, some of the Cu 2+ ions from the solution are dehydrated and transferred to the metal, creating a positive charge on it. This charge will prevent the further transfer of Cu 2+ ions from the solution to the metal and will lead to the formation of a layer of SO 4 2- anions attracted to it near the electrode. The so-called electrochemical equilibrium , at which the chemical potentials of the ions in the metal and in the solution will differ by the magnitude of the potential difference of the resulting double electric layer (DEL):
The electrical potential difference and the chemical potential difference are compensated in electrochemical equilibrium.
Let the concentration of CuSO 4 be so low that< . В этом случае при погружении металла в раствор будет наблюдаться обратный процесс перехода ионов меди из кристаллической решетки металла в раствор и электрод окажется заряженным отрицательно. Этот заряд будет препятствовать дальнейшему переходу ионов Cu 2+ в раствор, установится новое электрохимическое равновесие.
You can choose an electrolyte concentration at which the chemical potentials of the ions in the metal and the solution are the same. Solutions of this concentration are called zero solutions . When a metal is immersed in its zero solution, no EDL occurs on the surface of the electrode; however, even in this case, the potential difference between the metal and the solution is not zero.
According to Nernst, the only source of EMF of an electrochemical cell is the EMF on the surface of the electrodes. Nernst defined the potential of metals in a zero solution as absolute zero potential. In the works of A.N. Frumkin it was shown that Nernst’s ideas are incorrect. It has been experimentally established that the EMF of an element composed of two different electrodes immersed in their zero solutions differs very significantly from zero (maybe more than 1 V). The potential of a metal in a zero solution, called zero charge potential , cannot be considered as absolute zero potential.
THEORY OF CONDENSED HELMHOLTZ DOUBLE LAYER. The first quantitative theory of the structure of DEL at the metal-solution interface was created by Helmholtz (1853). According to Helmholtz, an EDL can be likened to a flat capacitor, one of the plates of which coincides with the plane passing through the surface charges in the metal, the other with the plane connecting the centers of the charges of ions in the solution, attracted to the surface of the metal by electrostatic forces. Double layer thickness l equal to the ion radius r. According to the condition of electrical neutrality, the number of ions attracted to the surface of the metal must be such that their charges compensate the surface charges of the metal, i.e.
The theory of a condensed double layer makes it possible to obtain values of the EDL capacitance that are consistent with experiment and a physically plausible thickness of the EDL. However, it cannot interpret many experimental regularities: the experimentally found values of the electrokinetic potential (x-potential) and their dependence on the electrolyte concentration, the change in the sign of the charge on the metal surface in the presence of a surfactant.
THEORY OF DIFFUSE DOUBLE LAYER GUI- CHAPMAN. Helmholtz's theory does not take into account that the properties of the DES change with the concentration of the electrolyte and T. Gouy (1910) and Chapman (1913) tried to relate the charge density in the DES with the composition of the solution. They took into account that in addition to the electrostatic forces that arise between the metal and the ions, the ions are also affected by the forces of thermal molecular motion. When these two forces are applied, the ions in the solution should be diffusely distributed relative to the metal surface - with a volumetric charge density decreasing with distance from it.
Gouy and Chapman believed that ions can be considered as material points that do not have their own volume, but have a charge, and that their distribution in the electrode charge field obeys the Boltzmann distribution.
The Gouy-Chapman theory agrees better than the Helmholtz theory with the laws of electrokinetic phenomena. If we assume that starting from a certain distance l 1 ions are no longer firmly bound to the electrode surface with relative movement of the solid and liquid phases, then the potential corresponding to this distance can be considered the x-potential (x< j). Однако теория не объясняет изменение знака x-потенциала и перезарядку поверхности с изменением состава раствора. Кроме того, теория Гуи-Чапмана оказывается менее удовлетворительной, чем теория Гельмгольца, при использовании ее для количественных расчетов емкости ДЭС, т.к. она не учитывает собственного объема ионов, которые отождествляются с материальными точками.
Thus, the Gouy-Chapman theory is best justified where the Helmholtz theory turns out to be inapplicable, and, conversely, the latter gives better convergence with experiment in cases where the first gives incorrect results. Consequently, the structure of the DES must correspond to some combination of the models proposed by Helmholtz and Gouy-Chapman. This assumption was made by Stern (1924) in his adsorption theory of DEL.
STERN ADSORPTION THEORY. Stern believed that a certain part of the ions is retained near the metal-electrolyte interface, forming a Helmholtz or condensed double-layer plate with a thickness corresponding to the average radius of the electrolyte ions. The remaining ions included in the EDL are distributed diffusely with a gradually decreasing charge density. For the diffuse part of the EDL, Stern, like Gouy, neglected the intrinsic sizes of the ions. In addition, Stern suggested that in the dense part of the EDL, ions are retained due to not only electrostatic forces, but also specific adsorption forces, i.e. forces of non-Coulomb origin. Therefore, in solutions containing surface-active ions, their number in the dense part of the EDL can exceed the charge of the metal surface by a certain amount, depending on the properties of the ions and the charge of the metal. Thus, according to Stern, two models of DES should be distinguished, one of which relates to solutions of surface-inactive electrolytes, the other to solutions containing specifically adsorbed ions.
In the adsorption theory the equality is also preserved:
Q M = q L = q 1 + q 2
The charge density on the solution side q L consists of two parts: the charge density in the Helmholtz layer q 1 and the charge density in the diffuse layer q 2 .
Stern's theory allows us to define the x-potential as the potential drop in the diffuse part of the EDL, where the strong bond between the metal and the ions has already been lost. With this definition, the x-potential should not coincide with the Nerst potential, as is observed experimentally. Stern's theory was able to explain the recharging of the surface of a solid body.
At an infinitesimal concentration, all charges in the solution are distributed diffusely, and the structure of the EDL is described by the Gouy-Chapman theory. On the contrary, in concentrated solutions the structure of DES approaches the model proposed by Helmholtz. In the region of average concentrations, where x is comparable in magnitude to RT/F, its dependence on concentration can be expressed by approximate equations:
for positive values x: x = B - ln With
for negative values of x: x = B¢ + ln With
Stern's theory gives a qualitatively correct picture of the DEL. The determination of capacitance using the Stern model is consistent with experience both in terms of the capacitance values and in the nature of its dependence on the electrode potential and solution concentration. But Stern's theory is not free from shortcomings. These include the impossibility of a quantitative description of capacitance curves, especially when moving away from the zero charge potential.
FURTHER DEVELOPMENT OF THE THEORY OF DES STANDING. Many attempts have been made to develop a theory of DES that is quantitatively consistent with experimental data (Rice, Frumkin et al., Bockris, Devanathan, Esin, Muller, Parsons, Ershler, etc.). The most widely accepted model is Graham (1947). According to Graham, the DES coating in the solution consists not of two, but of three parts. The first, counting from the surface of the metal, is called the internal Helmholtz plane; it contains only surface-active ions (the charge of the plane is q 1) or, if they are not in the solution, solvent molecules (q 1 = 0); its potential related to the solution is denoted by y 1. The next one, removed from the metal surface at a distance to which ions (their charge centers) can approach, is called the outer Helmholtz plane; its total charge is q 2, and the potential of the plane is y 2. Behind the outer Helmholtz plane there is a diffuse layer with a potential varying from y 2 to zero and with a charge density coinciding with q 2 .
Graham's model reflects the main features and characteristics of the metal-electrolyte DES structure. It allows you to calculate differential capacitance curves for any concentration of a given electrolyte if there is an experimental curve for at least one of its solutions. However, this model does not cover all aspects of the problem.
When an electric current passes through a solution, currents flow on the surface of the electrodes. electrochemical reactions, which are accompanied by the flow of electrons to or from the electrode. In reverse processes, electrochemical reactions occurring at the interfaces between conductors of the first and second kind lead to the generation of an electric current.
Electrochemical processes differ from conventional chemical reactions in a number of features.
A chemical reaction is possible only when reacting particles collide. When they come into contact, it becomes possible for electrons to transfer from one particle to another. Whether such a transition actually occurs depends on the energy of the particles and their mutual orientation. The activation energy depends on the nature of the chemical reaction, and for ionic reactions it is usually low. The electron transition path is very short, which is also a feature of the chemical reaction. Collisions of particles can occur at any points of the reaction space at different mutual positions, therefore electronic transitions can occur in arbitrary directions, i.e. Features of the chemical process are the randomness of collisions and the lack of directionality of electronic transitions. As a result, the energetic effects of chemical reactions appear primarily in the form of heat (a minor work of expansion is also possible).
In order for the energy changes corresponding to a chemical transformation to manifest themselves in the form of electrical energy, i.e. In order for the electrochemical process to proceed, it is necessary to change the reaction conditions.
Electrical energy is always associated with the passage of electric current, i.e. flow of electrons in a certain direction. Therefore, the reaction must be carried out in such a way that the electronic transitions are not random, but occur in one direction, and their path must be significantly larger than the atomic size. Therefore, in electrochemical processes, the transition of electrons from one participant to another must occur at a considerable distance, for which spatial separation of the reaction participants is necessary. However, spatial separation alone is not enough, as it will simply cause the reaction to stop.
To carry out the electrochemical process, additional conditions are necessary: electrons must be torn off from some particles and transferred to others in one common way. This can be achieved by replacing direct contact between the participants in the reaction with their contact with two metals connected to each other by a metal conductor. In order for the flow of electrons to be continuous, it is also necessary to ensure the passage of electric current through the reaction space, which is usually carried out by the participants in the electrochemical reaction themselves (if they are in an ionized state) or by special compounds with high ionic conductivity.
A device for producing electrical energy through electrochemical reactions is called electrochemical(or galvanic)element. The simplest electrochemical element consists of two metal electrodes (conductors of the first kind) immersed in an electrolyte solution (conductor of the second kind).
If, when an electric current passes in different directions, the same reaction occurs on the surface of the electrode, but in opposite directions, then such electrodes, as well as electrochemical elements composed of them, are called reversible. An example of a reversible element is the Daniel–Jacobi element
(–) Zn | ZnSO 4, solution || CuSO 4, solution | Cu(+)
When such an element operates, electrochemical reactions occur on the electrodes:
Zn Zn 2 + + 2e
Cu 2 + + 2eCu
The overall reaction equation in an element can be represented as
Zn + Cu 2 + Zn 2 + + Cu
When an infinitesimal current from an external source is passed through the element, these reactions proceed in the opposite direction.
Example irreversible element is the Volta element
(–) Zn | H2SO4 | Cu(+)
When such an element operates, the following reactions occur on the electrodes:
Zn Zn 2 + + 2e
2H + + 2eH 2 ,
and the reaction in the element is represented by the equation
Zn + 2H + Zn 2+ + H 2
When current is passed from an external source, other reactions occur on the electrodes:
Cu Cu 2 + + 2e,
those. in an electrochemical cell, copper dissolves in sulfuric acid with the release of hydrogen:
Cu + 2H + Cu 2 + + H 2
The most important characteristic of an electrochemical cell is its electromotive force(EMF) E– potential difference of a properly open element, i.e. the potential difference between the ends of conductors of the first kind of the same material connected to the electrodes of a galvanic cell. In other words, EMF is the potential difference under equilibrium conditions when no electric current flows in the circuit. If you close the electrodes, then an electric current will flow in the circuit, and the potential difference represents voltage an electrochemical element that differs from the EMF by the amount of voltage drop across the internal resistance of the element.
The purpose of electrochemical elements is to promote the movement of electrons in the external circuit in which the useful element is included
load. Thus, to perform its task, a fuel cell must contain a source and a sink of electrons (Fig. 7.1).
The reactions that occur in an electrochemical cell are called reduction-reaction, since the term “oxidation” corresponds to the process of releasing electrons, and the term “reduction” corresponds to the process of releasing electrons.
The meaning of numerous outdated scientific concepts is often not clear from their names. The terms “oxidation” and “reduction” require explanation. In Southeast, oxygen (oxygen) comes from the Latin root oxus (sour or hot, sharp) and means “acid producer.” This name was first used in the work of Morveau and Lavoisier “Nomenclature Chimique”, published in 1787. At that time, chemists adhered to an erroneous belief. that oxygen is the main element included in acids; when an acid is dissolved in water, part of the hydrogen atoms included in its composition loses its electron - the water becomes acidic, and the hydrogen is oxidized. Analystically, any reaction associated with the process of electron loss is called oxidation. The reverse reaction - the electron capture reaction - is called ■becoming.
Flow
J electrons
Electron source >
(oxidation)
Positive< -
direction of electric current Fig. 7.1. An electrochemical cell must contain a source and sink of electrons
In an electrochemical cell, the overall reaction is divided into two intermediate reactions that occur in separate areas of the device. These areas are fused by the electrolyte, which is a conductor of ions, but conducts electrons released in the intermediate oxidation reaction, electrons can enter the area where the reduction reaction occurs, but through an external circuit. Thus, an electric current arises in the external circuit, and the electrochemical cell is its source - this is its purpose. The positive direction of electric a in the external circuit was agreed to be the direction from the reducing oxidation region of the element - the electric current leaves the element of the reduction region, which is thus the cathode of the fuel - o the element, and enters the oxidation region, which is the anode. Like in any other electrical source, the cathode is positively charged
active electrode, and the anode is a negatively charged electrode. When it comes to consumers of electrical energy (load), the names change to the opposite. In the introduction to Ch. 6 The terms “anode” and “cathode” are discussed in more detail.
As an example of an electrochemical cell, consider a membrane that plays the role of an electrolyte. Let one of the side surfaces of the membranes be in contact with hydrogen. Under normal conditions the gas will consist predominantly of molecular hydrogen, but a small number of molecules may dissociate into atoms
and some of the atoms will oxidize (ionize), i.e. lose their electron
N -> Nt + e~ .
Since the membrane is impermeable to electrons, they will all remain on one side of it, while the formed ions through diff; through the membrane they will find themselves on the other side. In this case, the ions carry a positive charge, so the surface of the membrane on which hydrogen is located will become negatively charged due to the excess of electrons that have accumulated on it, and the opposite surface will become positively charged due to the positive ions that have appeared on it due to diffusion. The electric field causes some of the ions to move in the opposite direction to the “hydrogen” surface of the membrane. Dynamic equilibrium in the system under consideration is established when the diffusion flow of ions is st. i is equal to the reverse current.
Now let’s apply electrically conductive powder to both surfaces of the membrane. This will result in two porous electrically conductive layers that will act as electrodes. Let's connect an external heater1 to the electrodes, ensuring the electrical connection of the electrodes. Load resistance to significant Rl. Ions cannot move in the external circuit, and electrons begin to move from the “hydrogen” part, where there is an excess of them, to the opposite side of the membrane, forming an electric current in the external circuit, as in Fig. 7.2. The reaction of interest to us occurs at the “water” electrode and is described by the equation
2H2 -> 4H+ + 4e (anodic reaction)
The described scheme of operation of the element has one significant drawback: it contradicts the first law of thermodynamics. Indeed, when an electric current flows through an external load, the amount of it is released, the amount of which is determined by the product I2RL. At the same time, electric
entering the cathode region, combine with H+ ions, which diffuse through the membrane, forming hydrogen atoms, ultimately generating molecules of H2 gas used as “fuel”. If the process proceeded according to the given scheme, we would receive heat without any fuel consumption.
The external circuit only forms a path for the movement of electrons, but in itself it cannot be the cause of the occurrence of electrical current. Just like if you lower one end of a pipe into a lake, the water itself will not flow into the pipe. In order for water to flow, the other end must be below the surface of the water. Similarly, in order to introduce an electric current in an external circuit, it is necessary to reduce the rhmodynamic potential in the cathode region. The easiest way to do this is to add oxygen, the molecules of which combine with electrons and ions, resulting in the formation of water:
4е“ + 4Н+ + 02 -» 2Н20 (cathode reaction). (4)
The reaction is exothermic, i.e., it proceeds with the release of predominantly electrical energy, and not thermal energy, as happens when burning hydrogen. Obviously, it is this energy that powers the fuel element.
The described diagram of the electrochemical cell is shown in Fig. 7.2.
Under normal conditions, the fraction of dissipated hydrogen molecules * in the full region is small. It can be slightly increased by changing the physical parameters
meters in accordance with Le Chatelier's principle. for example, by increasing the system temperature. The degree of dissociation of hydrogen molecules can also be increased1 using the action of catalysts.
The complete reaction occurring in an electrochemical cell is described by the equation:
2H2 + 02 -> 2H?0.
The electrochemical cell, invented by Alessandro Volta (1745-1 in 1800), was the first device to generate electric current in a continuous mode. It consisted of zinc and silver (or copper) bases separated by sheets of paper soaked in a salt solution. The galvanic battery was formed By connecting the elements in series, the electrode of one element was connected directly to the ZINC electrode of another element.
An electrochemical Volta cell can be made by immersing welded and copper electrodes in a dilute (for example, 10%) solution of this acid. Zinc will oxidize:
Zn -> Zr,++ + 2e >
As a result, free electrons are formed. Dissolve zinc ions in water. Sulfuric acid, being a strong acid1), dissociates into ions:
H2S04 - "2Н+ + SOf"
Zinc ions combine with sulfate ions to form zinc sulfate. Pons in the form of the hydronium ion H" (H20)x will move through the electrolyte to the electrode, on which they will be released in the form of gas bubbles (when the hydronium ion captures electrons coming to the electrode from outside the II circuit). This type of electrochemical cell in practice almost useless, as very quickly the copper electrode will become covered with hydrogen bubbles adhering to its surface, which will significantly reduce the flow of ions entering the electrode. So-called "dry cells" use some method to prevent the formation of an insulating gas layer on the surface. The chemical reagent that is used to absorb the hydrogen is called a depolarizer. One of the reagents consumed is usually
"> The strength of an acid is determined by the degree of dissociation of its molecules in an aqueous solution. C acid completely dissociates into H+ and C1_ ions - this is a strong acid. Sulfuric acid is somewhat weaker, but also belongs to strong acids. Surprisingly, hydrofluoric acid, despite its high corrosiveness activity, is a weak acid - n t solution at room temperature, the proportion of H+ ions from the total concentration of neutra HF molecules is less than 3%.
gall, easily subject to oxidation; Zinc is most often used. Note that the copper electrode in the Volta element does not enter into chemical reactions, and therefore copper is not consumed.
Until recently, inexpensive galvanic batteries used Leclanchet cells, in which the anode is made of zinc and the cathode is made of a graphite rod coated with a layer of powdered manganese dioxide with the addition of graphite (to increase electrical conductivity). Manganese dioxide absorbs the released hydrogen, which prevents the formation of a gas layer on the cathode surface. Ammonium chloride is used as an electrolyte. Modern (alkaline) batteries use an alkaline electrolyte.
If the zinc used for the anode material were absolutely pure, it would only be consumed when electric current flows through the element. The presence of impurities causes corrosion of the electrode even when the element is not used (impurities form numerous microscopic electrochemical cells inside the electrode material). To ensure a long shelf life of such batteries, the anode is made of an alloy of zinc and mercury (amalgamated). Currently, Leclanche cells have been almost completely replaced by alkaline batteries.
Many chemical reactions occur only when energy is supplied from outside. They are often carried out in electrolytic cells (electrolyzers) on electrodes connected to an external current source. The study of these reactions provides information about the nature and properties of various substances, and also makes it possible to obtain new chemical compounds using electrosynthesis. Electrochemical processes are widely used in industry. Examples include chlorine and aluminum production, electroplating and electrical extraction. Galvanic cells, which convert chemical energy into electrical energy, form the basis of current sources - batteries and accumulators, as well as fuel cells. Electrochemistry also studies other electrical phenomena: the behavior of ions in electrolyte solutions and the passage of current through such solutions; separation of ions in an electric field (electrophoresis); corrosion and passivation of metals; electrical effects in biological systems (bioelectrochemistry); photoelectrochemical processes (the effect of light on electrochemical reactions in cells).
Historical reference.
It became possible to carry out systematic electrochemical research only after the creation of a constant, sufficiently powerful source of electric current. Such a source appeared at the turn of the 18th–19th centuries. as a result of the work of L. Galvani and A. Volta. While studying the physiological functions of the frog, Galvani accidentally created an electrochemical circuit consisting of two different metals and the muscle of a dissected frog leg. When the paw, secured with a copper holder, was touched with an iron wire, also connected to the holder, the muscle contracted. Similar contractions occurred under the influence of an electric discharge. Galvani explained this phenomenon by the existence of “animal electricity”. A different interpretation of these experiments was given by Volta, who believed that electricity arises at the point of contact of two metals, and the contraction of the frog muscle is the result of the passage of electric current through it. A current also occurred when a spongy material (cloth or paper) soaked in salt water was placed between two metal disks, for example zinc and copper, and the circuit was closed. By connecting 15–20 of these “elements” in series, Volta in 1800 created the first chemical current source - the “voltaic column”.
The influence of electricity on chemical systems immediately interested many scientists. Already in 1800, W. Nicholson and A. Carlyle reported that water decomposes into hydrogen and oxygen when an electric current is passed through it using platinum and gold wires connected to a “voltaic column.” The most important of the early electrochemical studies was the work of the English chemist H. Davy. In 1807, he isolated the element potassium by passing a current through slightly moistened solid potassium hydroxide. The voltage source was a battery of 100 galvanic cells. Metallic sodium was obtained in a similar way. Davy later isolated magnesium, calcium, strontium and barium by electrolysis using a mercury electrode.
Davy's assistant M. Faraday investigated the relationship between the amount of electricity (current times time) flowing through the electrode/solution interface and the chemical changes it caused. An instrument (now known as a gas coulometer) was created to measure the amount of electricity from the volume of hydrogen and oxygen released in an electrolytic cell, and it was shown (1833) that the amount of electricity required to obtain a given amount of substance does not depend on the size of the electrodes, the distance between them and the number of plates in the battery feeding the cell. In addition, Faraday discovered that the amount of a substance released during electrolysis is directly proportional to its chemical equivalent and the amount of electricity passed through the electrolyte. (A chemical equivalent is the number of grams of an element or compound that reacts with or replaces one mole of atoms (1.0078 g) of hydrogen in compounds; cm. EQUIVALENT MASS). These two fundamental provisions are called Faraday's laws. Together with his friend W. Whewell, a specialist in classical philology, Faraday also developed a new terminology in electrochemistry. He called conductors immersed in a solution electrodes (previously they were called poles); introduced the concepts of “electrolysis” (chemical changes associated with the passage of current), “electrolyte” (conducting liquid in electrochemical cells), “anode” (electrode on which the oxidation reaction occurs) and “cathode” (electrode on which the reduction reaction occurs ). He called charge carriers in liquids ions (from the Greek “wanderer”, “wanderer”), and the ions moving towards the anode (positive electrode) were called “anions”, and those moving towards the cathode - “cations”. Faraday's research on electromagnetic induction led to the creation of electrical generators, which made it possible to carry out electrochemical processes on an industrial scale.
Faraday explained the ability of solutions to pass electric current by the presence of ions in them, but he himself and other scientists, such as I. Hittorf and F. Kohlrausch, believed that ions appear under the influence of current. In 1884, S. Arrhenius suggested that in fact, ions are formed simply when salt is dissolved in water. The works of S. Arrhenius, J. Van't Hoff and W. Ostwald were an important milestone in the development of the theory of electrolytes and ideas about the physicochemical properties of solutions and their thermodynamics. The correspondence between theory and experimental data on ionic conductivity and equilibria in solution became more complete after P. Debye and E. Hückel took into account long-range electrostatic interactions between ions in 1923.
A major contribution to electrochemical thermodynamics and specifically to elucidating the nature of the electric potential (voltage) in an electrochemical cell and the balance between electrical, chemical and thermal energy was made by J. Gibbs and W. Nernst. The electrochemical potential is determined by the chemical energy of the processes occurring in the cell, but also depends on their speed (kinetics). Modeling of kinetic processes on electrodes was carried out by Y. Tafel (1905), J. Butler (1924), M. Volmer (1930), A. N. Frumkin (1930–1933).
Electrochemical cells.
An electrochemical cell usually consists of two half-cells, each of which is an electrode immersed in its own electrolyte. Electrodes are made of electrically conductive material (metal or carbon), or less often of a semiconductor. The charge carriers in the electrodes are electrons, and the charge carriers in the electrolyte are ions. An aqueous solution of table salt (sodium chloride NaCl), which is an electrolyte, contains charged particles: sodium cations Na + and chlorine anions Cl –. If you place such a solution in an electric field, the Na + ions will move to the negative pole, and the Cl – ions will move to the positive pole. Molten salts, such as NaCl, are also electrolytes. Electrolytes can also be solids, for example b- alumina (sodium polyaluminate) containing mobile sodium ions, or ion-exchange polymers.
The half-cells are separated by a partition, which does not interfere with the movement of ions, but prevents mixing of electrolytes. The role of such a partition can be performed by a salt bridge, a tube with an aqueous solution closed at both ends with glass wool, an ion exchange membrane, or a porous glass plate. Both electrodes of the electrolytic cell can be immersed in the same electrolyte.
There are two types of electrochemical cells: galvanic cells and electrolytic cells (electrolysers). In a galvanic cell, chemical reactions occur spontaneously at the electrode/electrolyte interface, and the electrodes are connected to each other by a conductor. Several galvanic cells connected in series form a battery - a chemical source of current. In an electrolytic cell, reactions at the electrode/electrolyte interface occur due to an external source of electrical energy; the latter is converted into chemical energy of reaction products occurring at the electrodes. The structure of the galvanic cell is shown in Fig. 1, and the electrolyzer - in Fig. 2. Note that the same cell, depending on the operating mode, can behave either as a galvanic cell or as an electrolyzer. Thus, a lead-acid car battery acts as a galvanic cell when used to start the engine (while discharging it), and as an electrolyser when charged from a car generator or charger.
A simple galvanic cell, created in 1836 by J. Daniel (Fig. 1), consists of two electrodes: zinc, immersed in an aqueous solution of zinc sulfate, and copper, immersed in an aqueous solution of copper (II) sulfate. Such an element is similar to copper-zinc pairs in a voltaic column. When the external circuit is closed, the zinc atoms on the surface of the zinc electrode are oxidized to ions with the release of electrons: Zn ® Zn 2+ + 2e – . These electrons move along the external circuit to the copper electrode and reduce copper ions to atoms: Cu 2+ + 2e – ® Cu. The flow of electrons in the external circuit is the current generated by the element. The overall reaction leading to a chemical transformation and the generation of electrical energy has the form
Exactly the same reaction occurs when metallic zinc is added to a solution of copper sulfate, but in this case the chemical energy is converted into thermal energy.
Electrochemical cells are often represented schematically by denoting the boundary between the electrode and the electrolyte with a vertical or slash (| or /) and the salt bridge with two slashes (//). So, the galvanic cell in Fig. 1 answer entry
where M is the molar concentration of the solution.
In the electrolytic cell shown in Fig. 2, the same reactions occur as in industrial electrolyzers for producing chlorine and alkali: the conversion of brine (a concentrated aqueous solution of sodium chloride) into chlorine and sodium hydroxide NaOH:
Chloride ions on the graphite electrode are oxidized to chlorine gas, and water on the iron electrode is reduced to hydrogen and hydroxide ions. Electrolytes remain electrically neutral due to the movement of sodium ions through a partition - an ion exchange membrane. The electrode at which oxidation occurs (zinc in Fig. 1 and graphite in Fig. 2) is called the anode, and the electrode at which reduction occurs is called the cathode.
Electrode potential.
The main electrical parameters of electrochemical cells are current (measured in amperes, A) and potential (measured in volts, V). The current strength is determined by the rate of electrode reactions, and the potential is determined by the chemical energy of the processes occurring in the cell. It is equal to energy (measured in joules, J) divided by the amount of electricity (measured in coulombs, C), i.e. 1 V = 1 J / Cl. Therefore, the potential of an element (electromotive force, emf) is a measure of the energy generated during the reactions occurring in it. If the external circuit is open, then no electrode reactions occur.
The potential of a galvanic cell with an open external circuit provides information about the thermodynamics of its reactions. The potential of the element shown in Fig. 1, at solution concentrations of 1 M and a temperature of 25 ° C - its standard potential E° – equal to 1.10 V. Corresponding energy, Gibbs thermodynamic potential, D G°, is given by
Where n– the number of electrons transferred during the reaction (in this case 2), F– Faraday number (96 485 C / mole). The potential of a galvanic cell is equal to the potential difference between its two half-cells, i.e. the difference in its electrode potentials. Electrode potentials are measured relative to the potential of the reference electrode, which is conventionally taken to be zero ( cm. table). By agreement, a normal hydrogen electrode (N.H.E.) was selected as the standard electrode; it is a platinum plate, which is coated with platinum black, saturated with hydrogen gas at a pressure of 1.01H 10 5 Pa (1 atm.), and immersed in a solution containing H + ions with thermodynamic activity a= 1. Schematically, this electrode can be represented as Pt/H 2 (1.01H 10 5 Pa)/H + ( a= 1). The reaction 2H + + 2e – ® H 2 occurs on it. To determine the standard potential of a Cu/Cu 2+ copper electrode, assemble the following galvanic cell:
and for the half-reaction Cu 2+ + 2e – ® Cu the measurement gives
Similarly, for the Zn/Zn 2+ half-cell, in which the reaction Zn 2+ + 2e – ® Zn occurs, we obtain
The difference between these two standard electrode potentials is equal to the standard potential of the Zn–Cu element.
In fact, the hydrogen electrode is rarely used in potentiometric measurements, since it represents an ideal system that is difficult to implement in practice. Much more often, more convenient and compact comparison electrodes of various types are used, having a specific, carefully measured potential value relative to the current. Usually they use a calomel electrode (CE), consisting of metallic mercury, mercury chloride (calomel) and a solution of potassium chloride: Hg /Hg 2 Cl 2 /KCl. The following reaction occurs at the electrode:
Potential k.e. depends on the concentration of mercury ions, and the latter depends on the concentration of the KCl solution. For saturated KCl solution E° (k.e.) av = 0.2412 V (n.e.) at 25° C.
|
STANDARD ELECTRODE POTENTIALS |
|
| Electrode reaction |
E o , V |
| Li + + e – ® Li | |
| Mg 2+ + 2e – ® Mg | |
| Al 3+ + 3e – ® Al | |
| Zn 2+ + 2e – ® Zn | |
| Cr 3+ + e – ® Cr 2+ | |
| 2H + + 2e – ® H 2 | |
| Cu 2+ + 2e – ® Cu | |
| Fe 3+ + e – ® Fe 2+ | |
| O 2 + 4H + + 4e – ® 2H 2 O | |
| Cl 2 + 2e – ® 2Cl – | |
| F 2 + 2e – ® 2F – | |
Note that the substance of some electrodes is not included in the equation of the corresponding reaction. Yes, reaction
actually occurs on a platinum electrode in a Pt/Fe 3+, Fe 2+ cell. The platinum electrode is inert and only provides contact with the electrolyte containing the oxidized and reduced forms of the element (in this case, divalent and trivalent iron ions). The platinum electrode plays the same role in N.V.E.
Tables of electrode potentials allow you to calculate the EMF of a galvanic cell based on its electrode potentials. They can also be used to predict whether a particular redox reaction will occur. We can talk about a standard electrode potential only when the activity of the components participating in the reaction is equal to 1, i.e. their concentration in solution is close to 1M. Electrode potential E depends on the concentration of oxidized and reduced forms in solution and is related to them and the standard potential E° Nernst equation. For a generalized reaction
ox + n e – = red
this equation has the form
Where R– universal gas constant, T– absolute temperature, and – activities of oxidized and reduced forms. The activities of pure solids and liquids are considered to be 1. At 25° C RT/F= 0.025 V. By measuring the electrode potentials relative to the potential of the reference electrode, it is possible to determine the concentrations of substances in the solution using equation (10); this method is called potentiometry.
Electrode reactions.
Potentiometric measurements are carried out under conditions when there is no current in the electrochemical cell. This means that no overall chemical changes occur in it, and the measured potential (equilibrium) is determined by the thermodynamics of the reactions. Under these conditions, factors such as the size and shape of the electrodes or the intensity of stirring the solution do not affect the measured potential. If current flows through an electrochemical cell, then the rate of electrode reactions depends not only on thermodynamic parameters, but also on the current strength in accordance with the equation
Where n– number of electrons participating in a given electrode reaction, F– Faraday number. In this case, the potential of the electrochemical cell depends on kinetic factors, as well as on the material from which the electrode is made, the size and shape of the electrode, the intensity of stirring the solution and many other factors. The internal resistance of the cell cannot be neglected. In addition to the potential difference at both electrode/electrolyte boundaries, a voltage drop occurs in the solution itself, due to its resistance. This voltage drop makes it difficult to study the effects associated with the reactions occurring at both electrodes. Usually, the reaction is studied on one electrode, which is called a working or indicator electrode, using a three-electrode cell (Fig. 3): the third electrode (for example, saturated calomel) is placed in the same compartment as the working one, as close as possible to it in order to reduce minimize the effect of ohmic voltage drop. By measuring the current through the working electrode as a function of the potential of this electrode relative to the reference electrode, the so-called polarization curve.
When an external current is passed, the electrode potential differs from the equilibrium one. This deviation is called polarization, and its magnitude is called overvoltage. Overvoltage depends on several factors that limit the rate of electrode reactions. Fast electrode reactions at a given current density (current strength per unit electrode surface) occur at potentials close to thermodynamic, and therefore at low overvoltage. Slow reactions are characterized by high overvoltage. The rates of electrode reactions, and hence the overvoltage, depend on the concentration of reagents, temperature, solvent, electrode material, method and speed of mass transfer, and current density. The total overvoltage can be decomposed into several components: concentration, activation and reaction.
Concentration overvoltage is caused by the fact that when a current passes, the concentration of the reacting ion on the surface of the electrode changes, since electroactive substances are consumed in this area and reaction products are formed. Let us consider the reduction of Cu 2+ on a copper electrode. Initially, the concentration of Cu 2+ in the solution is 1M. In the absence of current in the circuit, the potential of the copper electrode is close to the standard potential of the Cu/Cu 2+ pair, i.e. 0.34 V relative to N.E. [ cm. equation (6)]. As the cathode current passes, the concentration of Cu 2+ ions on the electrode surface decreases, and the cathode potential, in accordance with the Nernst equation (10), becomes increasingly negative. Fresh portions of the reagent arrive from the solution to the electrode in different ways: as a result of diffusion, convection, and migration. The higher the speed of these processes (for example, the more intense the mixing), the lower the concentration overvoltage. This component of the total overvoltage can often be calculated. If concentration polarization makes the main contribution to the total overvoltage (this means that the rate of the remaining stages of the electrode reaction is high), then the reaction is called reversible or Nernstian.
Activation overvoltage occurs as a result of the fact that electron transfer on the electrode surface does not occur instantly, but at a finite speed. Let us consider the generalized electrode reaction ox + n e – = red. To transfer electrons to oxidized compounds at a given rate (i.e., at a given current density), it is necessary to overcome an energy barrier called the activation energy of the electrode reaction. This energy is supplied by the applied potential. The relationship between current density and activation overvoltage is described by the Butler–Volmer and Tafel equations, which can be used to determine the kinetic parameters of electrode reactions. Many electrode reactions, such as the reduction of water to hydrogen and its oxidation to oxygen, occur slowly. The rate of electrode reaction can greatly depend on the material from which the electrode is made and the properties of its surface. For example, on a mercury electrode, the reduction of water to hydrogen is significantly difficult: it is characterized by a high hydrogen overvoltage. This reaction occurs much faster and with less overvoltage on platinum; It is for this reason that platinum is used in the hydrogen reference electrode. The rate of reactions also depends on the substances that are adsorbed or bound to the electrode surface; Thus, cyanide ion and a number of organic compounds reduce the rate of hydrogen evolution on the surface of the platinum electrode. At the same time, some surfactants can significantly increase the rate of electrode reaction - they are called electrocatalysts.
Reaction overvoltage occurs when the transfer of electrons at the electrode is coupled with a chemical reaction in the solution. Such a reaction can serve as a source of particles involved in electron transfer, and at the same time limit the rate of the entire electrode process. This is why it is so important to know the details of the mechanism (i.e., stages and intermediate states) of electrode reactions. In many cases, the starting material undergoes several transformations before becoming the final product at the electrode, as in the case of the reduction of oxygen to water, a process of great practical importance. The total reaction has the form
and consists of several stages, at one of which the oxygen–oxygen bond is broken. Due to this multi-stage reaction, the reaction at most electrodes is slow, and on an industrial scale it is carried out in the presence of electrocatalysts. The mechanism of electrode reactions is studied using electroanalytical methods described below. Often the course of a reaction changes when the composition of the solution and the nature of the solvent change. For example, the reduction of oxygen in acetonitrile, where there is a deficiency of protons, proceeds in accordance with a simple one-electron mechanism:
Electrochemical methods of analysis.
Various electrochemical methods have been developed for the qualitative and quantitative analysis of chemical substances, which are often also useful for determining the thermodynamic and kinetic parameters of electrode reactions and studying their mechanisms.
Conductometry
is based on measuring the electrical conductivity of a solution and is used to determine the concentration of salts, acids, bases, etc. In conductometric determinations, electrodes made of identical materials are usually used, and the conditions for their conduct are selected in such a way as to minimize the contribution of potential jumps at both electrode/electrolyte interfaces (for example, high-frequency alternating current is used). In this case, the main contribution to the measured cell potential is made by the ohmic voltage drop IR, Where R– solution resistance. The electrical conductivity of a one-component solution can be related to its concentration, and measuring the electrical conductivity of electrolytes of complex composition allows one to estimate the total ion content in the solution and is used, for example, in monitoring the quality of distilled or deionized water. In another type of conductometry - conductometric titration - a known reagent is added in portions to the analyzed solution and the change in electrical conductivity is monitored. The equivalence point, at which a sharp change in electrical conductivity is noted, is determined from a graph of the dependence of this value on the volume of added reagent.
Potentiometry
used to determine various physicochemical parameters based on data on the potential of a galvanic cell. The electrode potential in the absence of current in the electrochemical circuit, measured relative to the reference electrode, is related to the concentration of the solution by the Nernst equation. In potentiometric measurements, ion-selective electrodes are widely used that are sensitive primarily to one ion in solution: a glass electrode for measuring pH and electrodes for the selective determination of sodium, ammonium, fluorine, calcium, magnesium, etc. ions. The surface layer of the ion-selective electrode can include enzymes, and the result is a system that is sensitive to the appropriate substrate. Note that the potential of an ion-selective electrode is determined not by the transfer of electrons, as in the case of substances with electronic conductivity, but mainly by the transfer or exchange of ions. However, the Nernst equation, which relates the electrode potential to the logarithm of the concentration (or activity) of a substance in solution, is also applicable to such an electrode. In potentiometric titration, the reagent is added to the solution being analyzed in portions and the change in potential is monitored. The S-shaped curves characteristic of this type of titration allow one to determine the equivalence point and find thermodynamic parameters such as the equilibrium constant and standard potential.
Voltammetry.
All varieties of voltammetric methods use a working microelectrode with a surface area of 10–7–10–1 cm2. The current-voltage curves obtained with its help make it possible to identify dissolved substances, determine their concentration, and often thermodynamic and kinetic parameters. The first voltammetric method - polarography - was proposed in 1922 by the Czech chemist J. Heyrovsky. The working electrode in his setup was a dripping mercury electrode. Mercury has a high hydrogen overvoltage, so a mercury electrode is convenient for studying processes occurring at negative potentials. The electrode surface is constantly renewed during the measurement process, which eliminates contamination of the electrode. Voltammetric studies are also carried out using solid electrodes, such as platinum and carbon, and use processes that occur at positive potentials. In linear potential sweep voltammetry (chronoamperometry), the potential changes linearly with time and the solution is not stirred, so that mass transfer occurs solely due to diffusion. In cyclic voltammetry, repeated triangular voltage pulses are applied to an electrode. Substances formed in the ascending section of the cycle are studied in its descending section. This method is especially effective for studying the mechanism of electrode reactions by analyzing polarization curves at different potential sweep rates and different solution concentrations. There are other types of voltammetry - differential pulse and square wave - in which voltage pulses of different shapes are superimposed on a linearly increasing potential. These methods are widely used to determine small concentrations of substances in solution. If during a voltammetric measurement the solution is mixed, which means that mass transfer occurs simultaneously using convection and diffusion, then we speak of hydrodynamic voltammetry. In this case, it is convenient to use a rotating disk electrode, since the experimental current-voltage curves can be directly compared with the theoretical ones.
Amperometry.
The method is based on measuring the limiting diffusion current passing through a solution at a fixed voltage between the indicator electrode and the reference electrode. In amperometric titration, the equivalence point is determined by the break in the current curve - the volume of the added working solution. Chronoamperometric methods are based on measuring the dependence of current on time and are mainly used to determine diffusion coefficients and rate constants. Miniature electrochemical cells that serve as sensors at the output of liquid chromatograph columns operate on the principle of amperometry (as well as voltammetry). Galvanostatic methods are similar to amperometric ones, but they measure the potential when a certain amount of current passes through the cell. Thus, in chronopotentiometry, the change in potential over time is controlled. These methods are used mainly to study the kinetics of electrode reactions.
Coulometry.
In coulometry, at a controlled potential, complete electrolysis of a solution is carried out by intensively mixing it in an electrolyzer with a relatively large working electrode (bottom mercury or platinum mesh). Total amount of electricity ( Q, C) required for electrolysis is related to the amount of the forming substance ( A, d) Faraday's law:
Where M- they say mass (g/mol), F- Faraday number. Coulometric titration involves using a constant current to electrolytically generate a reagent that reacts with the substance being determined. The progress of the titration is controlled potentiometrically or amperometrically. Coulometric methods are convenient because they are absolute in nature (i.e., they allow you to calculate the amount of the analyte without resorting to calibration curves) and are insensitive to changes in electrolysis conditions and electrolyzer parameters (electrode surface area or stirring intensity). In coulogravimetry, the amount of substance that has undergone electrolysis is determined by weighing the electrode before and after electrolysis.
There are other electroanalytical methods. In alternating current polarography, a low amplitude sinusoidal voltage is applied to a linearly varying potential over a wide frequency range and either the amplitude and phase shift of the resulting alternating current or the impedance is determined. From these data, information is obtained about the nature of substances in solution and about the mechanism and kinetics of electrode reactions. Thin-layer methods use electrochemical cells with an electrolyte layer 10–100 µm thick. In such cells, electrolysis proceeds faster than in conventional electrolyzers. To study electrode processes, spectrochemical methods with spectrophotometric registration are used. To analyze substances formed on the surface of the electrode, their absorption of light in the visible, UV and IR regions is measured. Changes in the properties of the electrode surface and the medium are monitored using electrical reflection and ellipsometry methods, which are based on measuring the reflection of radiation from the electrode surface. These include methods of specular reflection and Raman scattering of light (Raman spectroscopy), second harmonic spectroscopy (Fourier spectroscopy).
Other electrochemical phenomena and methods.
With the relative movement of the electrolyte and charged particles or surfaces, electrokinetic effects occur. An important example of this kind is electrophoresis, in which the separation of charged particles (for example, protein molecules or colloidal particles) moving in an electric field occurs. Electrophoretic methods are widely used to separate proteins or deoxyribonucleic acids (DNA) in gels. Electrical phenomena play a large role in the functioning of living organisms: they are responsible for the generation and propagation of nerve impulses, the occurrence of transmembrane potentials, etc. Various electrochemical methods are used to study biological systems and their components. It is also of interest to study the effect of light on electrochemical processes. Thus, the subject of photoelectrochemical research is the generation of electrical energy and the initiation of chemical reactions under the influence of light, which is very important for increasing the efficiency of converting solar energy into electrical energy. Semiconductor electrodes made of titanium dioxide, cadmium sulfide, gallium arsenide and silicon are commonly used here. Another interesting phenomenon is electrochemiluminescence, i.e. generation of light in an electrochemical cell. It is observed when high-energy products are formed on the electrodes. Often the process is carried out in a cyclic manner to obtain both oxidized and reduced forms of a given compound. Their interaction with each other leads to the formation of excited molecules, which pass to the ground state with the emission of light.
Applied electrochemistry.
Electrochemistry has many practical applications. With the help of primary galvanic cells (disposable elements) connected to batteries, chemical energy is converted into electrical energy. Secondary current sources - batteries - store electrical energy. Fuel cells are primary power sources that generate electricity through a continuous supply of reactants (such as hydrogen and oxygen). These principles underlie portable power sources and batteries used on space stations, electric vehicles and electronic devices.
Large-scale production of many substances is based on electrochemical synthesis. Electrolysis of brine in the chlor-alkali process produces chlorine and alkali, which are then used to produce organic compounds and polymers, as well as in the pulp and paper industry. The products of electrolysis are compounds such as sodium chlorate, persulfate, sodium permanganate; Industrially important metals are obtained by electroextraction: aluminum, magnesium, lithium, sodium and titanium. It is better to use molten salts as electrolytes, since in this case, unlike aqueous solutions, the reduction of metals is not complicated by the release of hydrogen. Fluorine is produced by electrolysis in molten salt. Electrochemical processes serve as the basis for the synthesis of some organic compounds; for example, adiponitrile (an intermediate in the synthesis of nylon) is obtained by hydrodimerization of acrylonitrile.
Electroplating of silver, gold, chromium, brass, bronze and other metals and alloys is widely practiced on various objects in order to protect steel products from corrosion, for decorative purposes, for the manufacture of electrical connectors and printed circuit boards in the electronics industry. Electrochemical methods are used for high-precision dimensional processing of workpieces made of metals and alloys, especially those that cannot be processed by conventional mechanical methods, as well as for the manufacture of parts with complex profiles. When the surface of metals such as aluminum and titanium is anodized, protective oxide films are formed. Such films are created on the surface of workpieces made of aluminum, tantalum and niobium in the manufacture of electrolytic capacitors, and sometimes for decorative purposes.
Electrochemical methods are often used to base studies of corrosion processes and the selection of materials that slow down these processes. Corrosion of metal structures can be prevented using cathodic protection, for which an external source is connected to the structure being protected and the anode and the structure is maintained at a potential such that its oxidation is excluded. The possibilities of practical application of other electrochemical processes are being explored. So, electrolysis can be used to purify water. A very promising direction is the conversion of solar energy using photochemical methods. Electrochemical monitors are being developed, the operating principle of which is based on electrochemiluminescence. Let us also mention the study of reversible changes in the color of the electrode surface as a result of electrode reactions.
Literature:
Agladze R.I. Applied electrochemistry. M. – L., 1975
Izmailov N.A. Electrochemistry of solutions. M., 1976
Measurement methods in electrochemistry, vol. 1–2. M., 1977
Koryta I. Ions, electrodes, membranes. M., 1983
Bagotsky V.S. Basics of electrochemistry. M., 1988