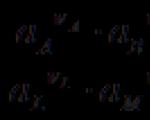Elettrochimica. Cella galvanica Cella elettrochimica
Elementi elettrochimici. Forza elettromotiva. Termodinamica di una cella galvanica. Misurazione dei campi elettromagnetici.
Doppio strato elettrico, meccanismo di occorrenza e struttura.
ELEMENTI GALVANICI. Campo elettromagnetico.
Quando una corrente elettrica passa attraverso un elettrolita, sulla superficie degli elettrodi si verificano reazioni elettrochimiche. Il verificarsi di reazioni elettrochimiche può essere generato da una fonte di corrente esterna. È possibile anche il fenomeno opposto: le reazioni elettrochimiche che avvengono su due elettrodi immersi in un elettrolita generano una corrente elettrica e le reazioni avvengono solo in un circuito chiuso (quando passa corrente).
Cella elettrochimica (o galvanica). è un dispositivo per produrre corrente elettrica attraverso reazioni elettrochimiche. L'elemento elettrochimico più semplice è costituito da due elettrodi metallici (conduttori del primo tipo), immersi in un elettrolita (conduttore del secondo tipo) e collegati tra loro da un contatto metallico. Diversi elementi elettrochimici collegati in serie circuito elettrochimico .
La caratteristica quantitativa più importante di un elemento elettrochimico è la forza elettromotrice(EMF, E), che è uguale alla differenza potenziale aprire correttamente l'elemento (quello in cui conduttori del primo tipo dello stesso materiale sono collegati agli elettrodi finali dell'elemento).
Se, quando una corrente elettrica passa in direzioni diverse, sulla superficie dell'elettrodo avviene la stessa reazione, ma in direzioni opposte, allora tali elettrodi, così come l'elemento o circuito da essi costituito, vengono chiamati reversibile . La fem degli elementi reversibili è la loro proprietà termodinamica, cioè dipende solo da T, P, dalla natura delle sostanze che compongono gli elettrodi e dalle soluzioni e dalla concentrazione di queste soluzioni. Un esempio di elemento reversibile è Elemento Daniel-Jacobi :
(-) Cu çZn çZnSO 4 ççCuSO 4 çCu (+)
in cui ciascun elettrodo è reversibile. Quando l'elemento funziona, si verificano le seguenti reazioni: Zn ® Zn 2+ + 2 e, Cu2+ + 2 e® Cu. Quando una corrente di intensità infinitesimale viene fatta passare da una sorgente esterna, sugli elettrodi si verificano reazioni inverse.
Un esempio di elemento irreversibile è Elemento volta :
(-) Zn ç H 2 SO 4 ç Cu (+)
Quando l'elemento funziona, si verificano le seguenti reazioni: Zn ® Zn 2+ + 2 e, 2H + + 2 e®H2. Quando si passa corrente da una fonte esterna, le reazioni degli elettrodi saranno: 2H + + 2 e® H 2 , Cu ® Cu 2+ + 2 e .
La forza elettromagnetica di un elemento elettrochimico è un valore positivo, perché corrisponde a un certo processo spontaneo che produce un lavoro positivo. Il processo inverso, che non può avvenire in maniera indipendente, corrisponderebbe ad un CEM negativo. Quando si compone una catena di elementi elettrochimici, il processo in uno degli elementi può essere diretto in modo tale da essere accompagnato dal dispendio di lavoro dall'esterno (processo non spontaneo), utilizzando per questo il lavoro di un altro elemento della catena in cui avviene un processo spontaneo. La fem totale di qualsiasi circuito è uguale alla somma algebrica delle quantità positive e negative. Pertanto, è molto importante quando si scrive uno schema elettrico tenere conto dei segni dell'EMF, utilizzando le regole accettate.
La fem del circuito elettrochimico è considerata positiva, se, durante la scrittura del circuito, l'elettrodo destro viene caricato positivamente rispetto a quello sinistro (durante il funzionamento del circuito, i cationi passano nella soluzione dall'elettrodo scritto a sinistra verso l'elettrodo scritto a destra e gli elettroni si muovono dentro stessa direzione nel circuito esterno). Esempio.
TERMODINAMICA DI UNA CELLA GALVANICA .
Lasciamo che la reazione proceda reversibilmente e isotermamente in un sistema elettrochimico:
n A A + n B B + ... ± nF Û n L L + n M M + ... ±
L'energia elettrica generata dall'elemento è pari al lavoro utile A¢ dell'intero processo. Il lavoro utile A¢ della trasformazione reversibile è massimo e in P, T = cost è pari alla diminuzione del potenziale isobarico del sistema:
DG P, T = nFE P, T
E P , T - EMF reversibile del sistema.
E P,T = -DG P,T / nF , E V,T = -DF V,T / nF
Pertanto, misurando la forza elettromagnetica dell'elemento e il suo coefficiente di temperatura, è possibile trovare i valori di DG e DS per l'intero processo che avviene nella cella galvanica. Questo processo è spontaneo, quindi DG< 0.
Utilizzando l'equazione di Gibbs-Helmholtz, possiamo calcolare la variazione di entalpia del processo:
DH = DG - T = -nFE P + TnF
nFE P = -DH + nFT = + nFT
nFE V = -DU + nFT = + nFT
Dalle equazioni ne consegue che la relazione tra l'energia elettrica generata o assorbita in modo reversibile in un sistema elettrochimico e l'effetto termico della reazione che si verifica in esso dipende dal segno e dall'entità del coefficiente di temperatura della fem dE/ dT :
1. SedE / dT > 0 , allora nFE > (DG > DH) e il sistema convertirà in energia elettrica non solo la quantità di calore che corrisponde all'effetto termico della reazione, ma anche calore aggiuntivo - Il calore di Peletier Q P = nFT dE/ dT preso in prestito dall’ambiente. In condizioni adiabatiche (in condizioni di isolamento termico, quando lo scambio con l'ambiente è impossibile), la T del sistema diminuisce. Il raffreddamento del sistema è particolarmente evidente se dE/ dT > 0 < 0 (реакция эндотермична).
2. SedE / dT < 0 , quindi nFE< (DG < DH) и часть теплоты реакции будет рассеиваться в виде теплоты Пелетье. В адиабатическом режиме система будет нагреваться.
3. SedE / dT = 0 , allora DG = DH e nFE = - l'energia elettrica prodotta reversibilmente dal sistema equivale all'effetto termico della reazione chimica. Questa relazione è nota come Principio di Thomson (regola) .
Per calcolare la FEM, le equazioni possono essere riscritte come:
Quando si usano le equazioni, è necessario ricordare che loro valido solo per sistemi elettrochimici reversibili, pertanto, quando si studia la dipendenza dei campi elettromagnetici da T, è necessario evitare l'uso di sistemi elettrochimici con confini liquidi, perché i potenziali di diffusione che si presentano su di essi non sono equilibrio.
Associamo la FEM dell'elemento alla costante di equilibrio della reazione che avviene nell'elemento. Equazione dell'isoterma della reazione chimica:
DG = RT ln K UN -RT
E = - = ln K UN -
Il primo termine sul lato destro dell'equazione per dati P, T è un valore costante; può essere indicato con E o. Eo- EMF standard di un elemento (sistema elettrochimico) , cioè. EMF affatto un io = 0.
E = Eo+ ln
 = Eo+2.303 lg
= Eo+2.303 lg
Pertanto, la forza elettromagnetica di un sistema elettrochimico è una funzione delle attività dei partecipanti alla reazione elettrochimica. Le equazioni di cui sopra consentono di calcolare i valori di DG e K UN basandosi sui valori sperimentali di E e, viceversa, calcolare E, conoscendo le caratteristiche termodinamiche della reazione chimica.
MISURAZIONE EMF .
Per misurare il valore di equilibrio (reversibile) della FEM di un elemento elettrochimico, è necessario che il processo avvenga in modo infinitamente lento, cioè in modo che l'elemento funzioni con una corrente infinitesimale. Questa condizione è soddisfatta nel metodo di compensazione, che si basa sul fatto che l'elemento è collegato in serie contro una differenza di potenziale esterna e quest'ultima è scelta in modo che non vi sia corrente nel circuito. Quindi la differenza di potenziale esterno è uguale alla FEM del circuito.
Utilizzando il metodo di compensazione, è possibile misurare direttamente il valore dell'EMF, ma questa è un'operazione piuttosto complicata, quindi nella pratica di laboratorio si preferisce confrontare l'EMF dell'elemento studiato con l'EMF del cosiddetto standard (normale) elementi, che viene attentamente misurato a T diverse. Questo metodo comparativo è anche una compensazione.
L'elemento normale di base è elemento Weston saturo .
(Circuito di misurazione EMF - indipendentemente).
STRUTTURA DEL CONFINE DELL'ELETTRODO - SOLUZIONE. DOPPIO STRATO ELETTRICO .
Quando un conduttore del primo tipo entra in contatto con un elettrolita, a doppio strato elettrico . Ad esempio, consideriamo un elettrodo di rame immerso in una soluzione di CuSO 4. Il potenziale chimico degli ioni rame in un metallo ad una data T può essere considerato costante, mentre il potenziale chimico degli ioni rame in soluzione dipende dalla concentrazione del sale; in generale, questi potenziali chimici non sono gli stessi.
Sia la concentrazione di CuSO 4 tale che > . Quindi, quando il metallo viene immerso nella soluzione, alcuni ioni Cu 2+ della soluzione vengono disidratati e trasferiti al metallo, creando su di esso una carica positiva. Questa carica impedirà l'ulteriore trasferimento di ioni Cu 2+ dalla soluzione al metallo e porterà alla formazione di uno strato di anioni SO 4 2- attratti ad esso vicino all'elettrodo. Il cosidetto equilibrio elettrochimico , al quale i potenziali chimici degli ioni nel metallo e nella soluzione differiranno dell'entità della differenza di potenziale del doppio strato elettrico risultante (DEL):
La differenza di potenziale elettrico e la differenza di potenziale chimico sono compensate nell'equilibrio elettrochimico.
Lascia che la concentrazione di CuSO 4 sia così bassa che< . В этом случае при погружении металла в раствор будет наблюдаться обратный процесс перехода ионов меди из кристаллической решетки металла в раствор и электрод окажется заряженным отрицательно. Этот заряд будет препятствовать дальнейшему переходу ионов Cu 2+ в раствор, установится новое электрохимическое равновесие.
È possibile scegliere una concentrazione dell'elettrolita alla quale i potenziali chimici degli ioni nel metallo e nella soluzione sono gli stessi. Vengono chiamate soluzioni di questa concentrazione soluzioni nulle . Quando un metallo è immerso nella sua soluzione nulla, sulla superficie dell'elettrodo non si forma alcun EDL; tuttavia, anche in questo caso, la differenza di potenziale tra il metallo e la soluzione non è nulla.
Secondo Nernst, l'unica fonte di campi elettromagnetici di una cella elettrochimica sono i campi elettromagnetici sulla superficie degli elettrodi. Nernst definì il potenziale dei metalli in una soluzione zero come potenziale zero assoluto. Nelle opere di A.N. Frumkin è stato dimostrato che le idee di Nernst sono errate. È stato stabilito sperimentalmente che la forza elettromagnetica di un elemento composto da due diversi elettrodi immersi nelle loro soluzioni zero differisce in modo molto significativo da zero (forse più di 1 V). Il potenziale di un metallo in una soluzione zero, chiamato potenziale di carica zero , non può essere considerato un potenziale zero assoluto.
TEORIA DEL DOPPIO STRATO CONDENSATO DI HELMHOLTZ. La prima teoria quantitativa della struttura del DEL all'interfaccia metallo-soluzione fu creata da Helmholtz (1853). Secondo Helmholtz, un EDL può essere paragonato a un condensatore piatto, una delle armature del quale coincide con il piano che passa attraverso le cariche superficiali del metallo, l'altra con il piano che collega i centri delle cariche degli ioni nella soluzione, attratto dalla superficie del metallo da forze elettrostatiche. Doppio spessore dello strato l uguale al raggio ionico R. Secondo la condizione di neutralità elettrica, il numero di ioni attratti dalla superficie del metallo deve essere tale che le loro cariche compensino le cariche superficiali del metallo, cioè
La teoria del doppio strato condensato consente di ottenere valori della capacità EDL coerenti con l'esperimento e uno spessore dell'EDL fisicamente plausibile. Tuttavia, non può interpretare molte regolarità sperimentali: i valori del potenziale elettrocinetico (potenziale x) riscontrati sperimentalmente e la loro dipendenza dalla concentrazione dell'elettrolita, il cambiamento del segno della carica sulla superficie metallica in presenza di un tensioattivo .
TEORIA DELLA GUI DIFFUSA A DOPPIO STRATO- CHAPMAN. La teoria di Helmholtz non tiene conto del fatto che le proprietà del DES cambiano con la concentrazione dell'elettrolita e T. Gouy (1910) e Chapman (1913) cercarono di mettere in relazione la densità di carica nel DES con la composizione della soluzione. Hanno tenuto conto del fatto che oltre alle forze elettrostatiche che si creano tra il metallo e gli ioni, gli ioni sono influenzati anche dalle forze del movimento termico molecolare. Quando vengono applicate queste due forze, gli ioni nella soluzione dovrebbero essere distribuiti diffusamente rispetto alla superficie metallica, con una densità di carica volumetrica decrescente con la distanza da essa.
Gouy e Chapman credevano che gli ioni potessero essere considerati come punti materiali che non hanno un proprio volume, ma hanno una carica, e che la loro distribuzione nel campo di carica dell'elettrodo obbedisce alla distribuzione di Boltzmann.
La teoria di Gouy-Chapman concorda meglio della teoria di Helmholtz con le leggi dei fenomeni elettrocinetici. Se assumiamo che parta da una certa distanza l 1 non sono più saldamente legati alla superficie dell'elettrodo con relativo movimento delle fasi solida e liquida, quindi il potenziale corrispondente a questa distanza può essere considerato il potenziale x (x< j). Однако теория не объясняет изменение знака x-потенциала и перезарядку поверхности с изменением состава раствора. Кроме того, теория Гуи-Чапмана оказывается менее удовлетворительной, чем теория Гельмгольца, при использовании ее для количественных расчетов емкости ДЭС, т.к. она не учитывает собственного объема ионов, которые отождествляются с материальными точками.
Pertanto, la teoria di Gouy-Chapman è meglio giustificata laddove la teoria di Helmholtz risulta inapplicabile e, al contrario, quest'ultima fornisce una migliore convergenza con l'esperimento nei casi in cui la prima fornisce risultati errati. Di conseguenza, la struttura del DES deve corrispondere ad una qualche combinazione dei modelli proposti da Helmholtz e Gouy-Chapman. Questa ipotesi fu fatta da Stern (1924) nella sua teoria dell'adsorbimento di DEL.
TEORIA DELL'ADSORBIMENTO STERN. Stern credeva che una certa parte degli ioni fosse trattenuta vicino all'interfaccia metallo-elettrolita, formando una piastra di Helmholtz o condensata a doppio strato con uno spessore corrispondente al raggio medio degli ioni dell'elettrolita. I restanti ioni inclusi nell'EDL sono distribuiti diffusamente con una densità di carica gradualmente decrescente. Per la parte diffusa dell'EDL, Stern, come Gouy, ha trascurato le dimensioni intrinseche degli ioni. Inoltre, Stern ha suggerito che nella parte densa dell'EDL, gli ioni vengono trattenuti non solo a causa delle forze elettrostatiche, ma anche delle forze di adsorbimento specifiche, ad es. forze di origine non coulombiana. Pertanto, nelle soluzioni contenenti ioni tensioattivi, il loro numero nella parte densa dell'EDL può superare di una certa quantità la carica della superficie metallica, a seconda delle proprietà degli ioni e della carica del metallo. Pertanto, secondo Stern, dovrebbero essere distinti due modelli di DES, uno dei quali si riferisce a soluzioni di elettroliti superficialmente inattivi, l'altro a soluzioni contenenti ioni specificatamente adsorbiti.
Anche nella teoria dell’adsorbimento viene preservata l’uguaglianza:
Q M = q L = q 1 + q 2
La densità di carica sul lato della soluzione q L è composta da due parti: la densità di carica nello strato di Helmholtz q 1 e la densità di carica nello strato diffuso q 2 .
La teoria di Stern ci permette di definire il potenziale x come la caduta di potenziale nella parte diffusa dell'EDL, dove il forte legame tra il metallo e gli ioni è già andato perduto. Con questa definizione il potenziale x non dovrebbe coincidere con il potenziale di Nerst, come si osserva sperimentalmente. La teoria di Stern era in grado di spiegare la ricarica della superficie di un corpo solido.
A una concentrazione infinitesimale, tutte le cariche nella soluzione sono distribuite diffusamente e la struttura dell'EDL è descritta dalla teoria di Gouy-Chapman. Al contrario, nelle soluzioni concentrate la struttura del DES si avvicina al modello proposto da Helmholtz. Nella regione delle concentrazioni medie, dove x è paragonabile in grandezza a RT/F, la sua dipendenza dalla concentrazione può essere espressa mediante equazioni approssimative:
per valori positivi x: x = B - ln Con
per valori negativi di x: x = B¢+ ln Con
La teoria di Stern fornisce un'immagine qualitativamente corretta del DEL. La determinazione della capacità utilizzando il modello Stern è coerente con l'esperienza sia in termini di valori di capacità che nella natura della sua dipendenza dal potenziale dell'elettrodo e dalla concentrazione della soluzione. Ma la teoria di Stern non è esente da difetti. Questi includono l'impossibilità di una descrizione quantitativa delle curve di capacità, soprattutto quando ci si allontana dal potenziale di carica zero.
ULTERIORE SVILUPPO DELLA TEORIA DEL DES STANDING. Sono stati fatti molti tentativi per sviluppare una teoria dei DES che sia quantitativamente coerente con i dati sperimentali (Rice, Frumkin et al., Bockris, Devanathan, Esin, Muller, Parsons, Ershler, ecc.). Il modello più ampiamente accettato è Graham (1947). Secondo Graham, il rivestimento DES nella soluzione non è costituito da due, ma da tre parti. Il primo, partendo dalla superficie del metallo, è chiamato piano interno di Helmholtz; contiene solo ioni tensioattivi (la carica del piano è q 1) o, se non sono presenti in soluzione, molecole di solvente (q 1 = 0); il suo potenziale relativo alla soluzione è indicato con y 1. Il successivo, rimosso dalla superficie metallica ad una distanza alla quale possono avvicinarsi gli ioni (i loro centri di carica), è chiamato piano di Helmholtz esterno; la sua carica totale è q 2 e il potenziale del piano è y 2. Dietro il piano di Helmholtz esterno si trova uno strato diffuso con potenziale variabile da y 2 a zero e con densità di carica coincidente con q 2 .
Il modello di Graham riflette le principali caratteristiche e caratteristiche della struttura DES metallo-elettrolita. Permette di calcolare curve di capacità differenziale per qualsiasi concentrazione di un dato elettrolita se esiste una curva sperimentale per almeno una delle sue soluzioni. Tuttavia, questo modello non copre tutti gli aspetti del problema.
Quando una corrente elettrica passa attraverso una soluzione, le correnti fluiscono sulla superficie degli elettrodi. reazioni elettrochimiche, che sono accompagnati dal flusso di elettroni verso o dall'elettrodo. Nei processi inversi, le reazioni elettrochimiche che si verificano all'interfaccia tra conduttori del primo e del secondo tipo portano alla generazione di corrente elettrica.
I processi elettrochimici differiscono dalle reazioni chimiche convenzionali in una serie di caratteristiche.
Una reazione chimica è possibile solo quando le particelle reagenti entrano in collisione. Quando entrano in contatto, diventa possibile per gli elettroni trasferirsi da una particella all'altra. Il fatto che tale transizione avvenga effettivamente dipende dall'energia delle particelle e dal loro orientamento reciproco. L'energia di attivazione dipende dalla natura della reazione chimica e per le reazioni ioniche è generalmente bassa. Il percorso di transizione degli elettroni è molto breve, caratteristica anche della reazione chimica. Le collisioni di particelle possono verificarsi in qualsiasi punto dello spazio di reazione in diverse posizioni reciproche, pertanto le transizioni elettroniche possono verificarsi in direzioni arbitrarie, ad es. Caratteristiche del processo chimico sono la casualità delle collisioni e la mancanza di direzionalità delle transizioni elettroniche. Di conseguenza, gli effetti energetici delle reazioni chimiche si manifestano principalmente sotto forma di calore (è possibile anche un piccolo lavoro di espansione).
Affinché le variazioni energetiche corrispondenti ad una trasformazione chimica si manifestino sotto forma di energia elettrica, cioè Affinché il processo elettrochimico possa procedere è necessario modificare le condizioni di reazione.
L’energia elettrica è sempre associata al passaggio di corrente elettrica, cioè flusso di elettroni in una certa direzione. Pertanto, la reazione deve essere eseguita in modo tale che le transizioni elettroniche non siano casuali, ma avvengano in una direzione e il loro percorso sia significativamente più grande della dimensione atomica. Pertanto, nei processi elettrochimici, la transizione degli elettroni da un partecipante all'altro deve avvenire a una distanza considerevole, per la quale è necessaria la separazione spaziale dei partecipanti alla reazione. Tuttavia, la separazione spaziale da sola non è sufficiente, poiché causerebbe semplicemente l’arresto della reazione.
Per eseguire il processo elettrochimico sono necessarie condizioni aggiuntive: gli elettroni devono essere strappati da alcune particelle e trasferiti ad altre in un modo comune. Ciò può essere ottenuto sostituendo il contatto diretto tra i partecipanti alla reazione con il loro contatto con due metalli collegati tra loro da un conduttore metallico. Affinché il flusso di elettroni sia continuo, è anche necessario garantire il passaggio della corrente elettrica attraverso lo spazio di reazione, che di solito viene effettuato dagli stessi partecipanti alla reazione elettrochimica (se sono in uno stato ionizzato) o da speciali composti ad elevata conducibilità ionica.
Viene chiamato un dispositivo per produrre energia elettrica attraverso reazioni elettrochimiche elettrochimico(O galvanico)elemento. L'elemento elettrochimico più semplice è costituito da due elettrodi metallici (conduttori del primo tipo) immersi in una soluzione elettrolitica (conduttore del secondo tipo).
Se, quando una corrente elettrica passa in direzioni diverse, si verifica la stessa reazione sulla superficie dell'elettrodo, ma in direzioni opposte, allora vengono chiamati tali elettrodi, così come gli elementi elettrochimici da essi composti reversibile. Un esempio di elemento reversibile è l'elemento Daniel-Jacobi
(–) Zn | ZnSO 4, soluzione || CuSO4, soluzione | Cu(+)
Quando un tale elemento funziona, si verificano reazioni elettrochimiche sugli elettrodi:
Zn Zn 2 + + 2e
Cu 2 + + 2e Cu
L'equazione di reazione complessiva in un elemento può essere rappresentata come
Zn + Cu2 + Zn2 + + Cu
Quando una corrente infinitesima proveniente da una sorgente esterna viene fatta passare attraverso l'elemento, queste reazioni procedono nella direzione opposta.
Esempio irreversibile l'elemento è l'elemento Volta
(–) Zn | H2SO4 | Cu(+)
Quando un tale elemento funziona, sugli elettrodi si verificano le seguenti reazioni:
Zn Zn 2 + + 2e
2H + + 2eH 2 ,
e la reazione nell'elemento è rappresentata dall'equazione
Zn + 2H + Zn 2+ + H 2
Quando la corrente viene trasmessa da una fonte esterna, sugli elettrodi si verificano altre reazioni:
Cu Cu 2 + + 2e,
quelli. in una cella elettrochimica, il rame si dissolve in acido solforico con rilascio di idrogeno:
Cu + 2H + Cu 2 + + H 2
La caratteristica più importante di una cella elettrochimica è la sua forza elettromotiva(EMF) E– differenza di potenziale di un elemento propriamente aperto, cioè la differenza di potenziale tra le estremità di conduttori del primo tipo dello stesso materiale collegati agli elettrodi di una cella galvanica. In altre parole, la FEM è la differenza di potenziale in condizioni di equilibrio quando nel circuito non scorre corrente elettrica. Se chiudi gli elettrodi, nel circuito scorrerà corrente elettrica e la differenza di potenziale rappresenta voltaggio un elemento elettrochimico che differisce dall'EMF per la quantità di caduta di tensione attraverso la resistenza interna dell'elemento.
Lo scopo degli elementi elettrochimici è favorire il movimento degli elettroni nel circuito esterno in cui è incluso l'elemento utile
carico. Pertanto, per svolgere il suo compito, una cella a combustibile deve contenere una sorgente e un pozzo di elettroni (Fig. 7.1).
Le reazioni che si verificano in una cella elettrochimica sono chiamate reazione di riduzione, poiché il termine "ossidazione" corrisponde al processo di rilascio di elettroni e il termine "riduzione" corrisponde al processo di rilascio di elettroni.
Il significato di numerosi concetti scientifici obsoleti spesso non è chiaro dai loro nomi, i termini “ossidazione” e “riduzione” richiedono una spiegazione. Nel sud-est, l'ossigeno deriva dalla radice latina oxus (aspro o caldo, piccante) e significa "produttore di acido". Questo nome fu usato per la prima volta nell'opera di Morveau e Lavoisier “Nomenclature Chimique”, pubblicata nel 1787. A quel tempo, i chimici aderivano a una convinzione errata. che l'ossigeno è l'elemento principale incluso negli acidi; quando un acido viene sciolto in acqua, parte degli atomi di idrogeno inclusi nella sua composizione perde il suo elettrone: l'acqua diventa acida e l'idrogeno si ossida. Dal punto di vista analitico, qualsiasi reazione associata al processo di perdita di elettroni è chiamata ossidazione. La reazione inversa – la reazione di cattura degli elettroni – è chiamata ■divenire.
Fluire
Elettroni J
Sorgente di elettroni >
(ossidazione)
Positivo< -
direzione della corrente elettrica Fig. 7.1. Una cella elettrochimica deve contenere una sorgente e un pozzo di elettroni
In una cella elettrochimica, la reazione complessiva è divisa in due reazioni intermedie che avvengono in aree separate del dispositivo. Queste aree sono fuse dall'elettrolita, che è un conduttore di ioni, ma conduce gli elettroni rilasciati nella reazione di ossidazione intermedia, gli elettroni possono entrare nell'area in cui avviene la reazione di riduzione, ma attraverso un circuito esterno. Pertanto, nel circuito esterno si forma una corrente elettrica e la cella elettrochimica è la sua fonte: questo è il suo scopo. Si è convenuto che la direzione positiva dell'a elettrico nel circuito esterno sia la direzione dalla regione di ossidazione riducente dell'elemento - la corrente elettrica lascia l'elemento della regione di riduzione, che è quindi il catodo del combustibile - o l'elemento, e entra nella regione di ossidazione, che è l'anodo. Come in qualsiasi altra fonte elettrica, il catodo è carico positivamente
elettrodo attivo e l'anodo è un elettrodo caricato negativamente. Quando si tratta dei consumatori di energia elettrica (carico), i nomi cambiano al contrario. Nell'introduzione al cap. 6 I termini “anodo” e “catodo” verranno discussi più in dettaglio.
Come esempio di cella elettrochimica, considera una membrana che svolge il ruolo di elettrolita. Lasciare che una delle superfici laterali delle membrane sia a contatto con l'idrogeno. In condizioni normali il gas sarà costituito prevalentemente da idrogeno molecolare, ma un piccolo numero di molecole potrebbe dissociarsi in atomi
e alcuni atomi si ossideranno (ionizzeranno), cioè perderanno il loro elettrone
N -> Nt + e~ .
Poiché la membrana è impermeabile agli elettroni, essi rimarranno tutti su un lato della stessa, mentre gli ioni formatisi attraverso diff; attraverso la membrana si ritroveranno dall'altra parte. In questo caso, gli ioni trasportano una carica positiva, quindi la superficie della membrana su cui si trova l'idrogeno si caricherà negativamente a causa dell'eccesso di elettroni che si sono accumulati su di essa, e la superficie opposta si caricherà positivamente a causa della carica positiva ioni che sono apparsi su di esso a causa della diffusione. Il campo elettrico fa sì che alcuni ioni si muovano nella direzione opposta alla superficie “idrogeno” della membrana. L'equilibrio dinamico nel sistema in esame viene stabilito quando il flusso di diffusione degli ioni è st. i è uguale alla corrente inversa.
Ora applichiamo la polvere elettricamente conduttiva su entrambe le superfici della membrana: ciò si tradurrà in due strati porosi elettricamente conduttivi che fungeranno da elettrodi. Colleghiamo un riscaldatore esterno1 agli elettrodi, garantendo il collegamento elettrico degli elettrodi. Resistenza al carico fino a Rl significativo. Gli ioni non possono muoversi nel circuito esterno e gli elettroni iniziano a spostarsi dalla parte "idrogeno", dove ce n'è in eccesso, al lato opposto della membrana, formando una corrente elettrica nel circuito esterno, come in Fig. 7.2. La reazione che ci interessa avviene sull'elettrodo “acqua” ed è descritta dall'equazione
2H2 -> 4H+ + 4e (reazione anodica)
Lo schema di funzionamento descritto dell'elemento presenta uno svantaggio significativo: contraddice la prima legge della termodinamica. Infatti, quando una corrente elettrica attraversa un carico esterno, ne viene rilasciata una quantità, la cui quantità è determinata dal prodotto I2RL. Allo stesso tempo, elettrico
dirigendosi verso la regione del catodo, si combinano con gli ioni H+, che si diffondono attraverso la membrana, formando atomi di idrogeno, generando infine molecole di gas H2 utilizzato come “carburante”. Se il processo procedesse secondo lo schema indicato, riceveremmo calore senza alcun consumo di carburante.
Il circuito esterno costituisce solo un percorso per il movimento degli elettroni, ma di per sé non può essere la causa della formazione di corrente elettrica. Proprio come se abbassassi un'estremità di un tubo in un lago, l'acqua stessa non scorrerà nel tubo. Affinché l'acqua possa scorrere, l'altra estremità deve trovarsi sotto la superficie dell'acqua. Allo stesso modo, per introdurre una corrente elettrica in un circuito esterno, è necessario ridurre il potenziale remodinamico nella regione del catodo. Il modo più semplice per farlo è aggiungere ossigeno, le cui molecole si combinano con elettroni e ioni, dando luogo alla formazione di acqua:
4е“ + 4Н+ + 02 -» 2Н20 (reazione catodica). (4)
La reazione è esotermica, procede cioè con liberazione di energia prevalentemente elettrica e non termica, come avviene quando si brucia l'idrogeno. Ovviamente, è questa energia che alimenta l’elemento combustibile.
Lo schema descritto della cella elettrochimica è mostrato in Fig. 7.2.
In condizioni normali, la frazione di molecole di idrogeno dissipate * nell'intera regione è piccola. Può essere leggermente aumentato modificando i parametri fisici
metri secondo il principio di Le Chatelier. ad esempio, aumentando la temperatura del sistema. Il grado di dissociazione delle molecole di idrogeno può anche essere aumentato1 utilizzando l'azione dei catalizzatori.
La reazione completa che avviene in una cella elettrochimica è descritta dall'equazione:
2H2 + 02 -> 2H?0.
La cella elettrochimica, inventata da Alessandro Volta (1745-1 nel 1800), fu il primo apparecchio per generare corrente elettrica in modo continuo: era costituita da basi di zinco e argento (o rame) separate da fogli di carta imbevuti in una soluzione salina. Si formò la batteria galvanica Collegando gli elementi in serie, l'elettrodo di un elemento era collegato direttamente all'elettrodo di ZINCO di un altro elemento.
Una cella elettrochimica Volta può essere realizzata immergendo elettrodi saldati e di rame in una soluzione diluita (ad esempio al 10%) di questo acido. Lo zinco si ossiderà:
Zn -> Zr,++ + 2e >
Di conseguenza, si formano elettroni liberi. Sciogliere gli ioni di zinco in acqua. L'acido solforico, essendo un acido forte1), si dissocia in ioni:
H2S04 - "2Í+ + SOf"
Gli ioni zinco si combinano con gli ioni solfato per formare solfato di zinco. I ponti sotto forma di ione idronio H" (H20)x si sposteranno attraverso l'elettrolita fino all'elettrodo, sul quale verranno rilasciati sotto forma di bolle di gas (quando lo ione idronio cattura gli elettroni che arrivano all'elettrodo dall'esterno dell'II Questo tipo di cella elettrochimica in pratica è quasi inutile, poiché molto rapidamente l'elettrodo di rame si ricoprirà di bolle di idrogeno aderenti alla sua superficie, il che ridurrà significativamente il flusso di ioni che entrano nell'elettrodo. Le cosiddette "celle a secco" utilizzano un metodo per prevenire la formazione di uno strato di gas isolante sulla superficie. Il reagente chimico utilizzato per assorbire l'idrogeno è chiamato depolarizzatore. Uno dei reagenti consumati viene solitamente
"> La forza di un acido è determinata dal grado di dissociazione delle sue molecole in una soluzione acquosa. L'acido C si dissocia completamente negli ioni H+ e C1_: questo è un acido forte. L'acido solforico è leggermente più debole, ma appartiene anche agli acidi forti Sorprendentemente, l'acido fluoridrico, nonostante la sua elevata attività corrosiva, è una soluzione acido-n debole a temperatura ambiente, la proporzione di ioni H+ dalla concentrazione totale di molecole HF neutre è inferiore al 3%.
fiele, facilmente soggetto ad ossidazione; Lo zinco è più spesso usato. Si noti che l'elettrodo di rame nell'elemento Volta non entra in reazioni chimiche e quindi il rame non viene consumato.
Fino a poco tempo fa, le batterie galvaniche economiche utilizzavano celle Leclanchet, in cui l'anodo è di zinco e il catodo è costituito da un'asta di grafite rivestita con uno strato di biossido di manganese in polvere con l'aggiunta di grafite (per aumentare la conduttività elettrica). Il biossido di manganese assorbe l'idrogeno rilasciato, impedendo la formazione di uno strato di gas sulla superficie del catodo. Come elettrolita viene utilizzato il cloruro di ammonio. Le batterie moderne (alcaline) utilizzano un elettrolita alcalino.
Se lo zinco utilizzato per il materiale dell'anodo fosse assolutamente puro, verrebbe consumato solo quando la corrente elettrica scorre attraverso l'elemento. La presenza di impurità provoca la corrosione dell'elettrodo anche quando l'elemento non viene utilizzato (le impurità formano numerose cellule elettrochimiche microscopiche all'interno del materiale dell'elettrodo). Per garantire una lunga durata di tali batterie, l'anodo è costituito da una lega di zinco e mercurio (amalgamati). Attualmente le celle Leclanche sono state quasi completamente sostituite da batterie alcaline.
Molte reazioni chimiche avvengono solo quando l'energia viene fornita dall'esterno. Vengono spesso eseguiti in celle elettrolitiche (elettrolizzatori) su elettrodi collegati a una fonte di corrente esterna. Lo studio di queste reazioni fornisce informazioni sulla natura e le proprietà di varie sostanze e consente inoltre di ottenere nuovi composti chimici mediante elettrosintesi. I processi elettrochimici sono ampiamente utilizzati nell’industria. Gli esempi includono la produzione di cloro e alluminio, la galvanica e l'estrazione elettrica. Le celle galvaniche, che convertono l'energia chimica in energia elettrica, costituiscono la base delle fonti attuali: batterie e accumulatori, nonché celle a combustibile. L'elettrochimica studia anche altri fenomeni elettrici: il comportamento degli ioni nelle soluzioni elettrolitiche e il passaggio della corrente attraverso tali soluzioni; separazione di ioni in un campo elettrico (elettroforesi); corrosione e passivazione dei metalli; effetti elettrici nei sistemi biologici (bioelettrochimica); processi fotoelettrochimici (l'effetto della luce sulle reazioni elettrochimiche nelle cellule).
Riferimento storico.
È diventato possibile effettuare ricerche elettrochimiche sistematiche solo dopo la creazione di una fonte di corrente elettrica costante e sufficientemente potente. Tale fonte apparve a cavallo tra il XVIII e il XIX secolo. per opera di L. Galvani e A. Volta. Mentre studiava le funzioni fisiologiche della rana, Galvani creò accidentalmente un circuito elettrochimico costituito da due metalli diversi e dal muscolo di una coscia di rana sezionata. Quando la zampa, fissata con un supporto di rame, veniva toccata con un filo di ferro, anch'esso collegato al supporto, il muscolo si contraeva. Contrazioni simili si sono verificate sotto l'influenza di una scarica elettrica. Galvani spiegò questo fenomeno con l'esistenza dell'“elettricità animale”. Una diversa interpretazione di questi esperimenti fu data da Volta, il quale credeva che l'elettricità nascesse nel punto di contatto di due metalli, e la contrazione del muscolo della rana è il risultato del passaggio della corrente elettrica attraverso di esso. Una corrente si verificava anche quando un materiale spugnoso (stoffa o carta) imbevuto di acqua salata veniva posto tra due dischi metallici, ad esempio zinco e rame, e il circuito veniva chiuso. Collegando 15-20 di questi "elementi" in serie, Volta nel 1800 creò la prima sorgente di corrente chimica: la "colonna voltaica".
L'influenza dell'elettricità sui sistemi chimici interessò immediatamente molti scienziati. Già nel 1800, W. Nicholson e A. Carlyle riferirono che l’acqua si decompone in idrogeno e ossigeno quando viene attraversata da corrente elettrica utilizzando fili di platino e oro collegati a una “colonna voltaica”. Il più importante dei primi studi elettrochimici fu il lavoro del chimico inglese H. Davy. Nel 1807 isolò l'elemento potassio facendo passare una corrente attraverso idrossido di potassio solido leggermente inumidito. La sorgente di tensione era una batteria di 100 celle galvaniche. Il sodio metallico è stato ottenuto in modo simile. Davy successivamente isolò magnesio, calcio, stronzio e bario mediante elettrolisi utilizzando un elettrodo di mercurio.
L'assistente di Davy, M. Faraday, ha studiato la relazione tra la quantità di elettricità (corrente per tempo) che scorre attraverso l'interfaccia elettrodo/soluzione e i cambiamenti chimici che ha causato. Fu creato uno strumento (ora noto come coulometro a gas) per misurare la quantità di elettricità dal volume di idrogeno e ossigeno rilasciato in una cella elettrolitica e fu dimostrato (1833) che la quantità di elettricità necessaria per ottenere una data quantità di La sostanza non dipende dalla dimensione degli elettrodi, dalla distanza tra loro e dal numero di piastre nella batteria che alimenta la cella. Inoltre, Faraday scoprì che la quantità di sostanza rilasciata durante l'elettrolisi è direttamente proporzionale al suo equivalente chimico e alla quantità di elettricità che passa attraverso l'elettrolita. (Un equivalente chimico è il numero di grammi di un elemento o composto che reagisce con o sostituisce una mole di atomi (1,0078 g) di idrogeno nei composti; cm. MASSA EQUIVALENTE). Queste due disposizioni fondamentali sono chiamate leggi di Faraday. Insieme al suo amico W. Whewell, specialista in filologia classica, Faraday sviluppò anche una nuova terminologia in elettrochimica. Chiamò elettrodi i conduttori immersi in una soluzione (prima si chiamavano poli); introdotti i concetti di “elettrolisi” (cambiamenti chimici legati al passaggio di corrente), “elettrolita” (liquido conduttore nelle celle elettrochimiche), “anodo” (elettrodo su cui avviene la reazione di ossidazione) e “catodo” (elettrodo su cui avviene la avviene la reazione di riduzione). Chiamò i portatori di carica nei liquidi ioni (dal greco "vagabondo", "vagabondo"), e gli ioni che si muovevano verso l'anodo (elettrodo positivo) furono chiamati "anioni", e quelli che si muovevano verso il catodo - "cationi". Le ricerche di Faraday sull'induzione elettromagnetica portarono alla creazione di generatori elettrici, che permisero di eseguire processi elettrochimici su scala industriale.
Faraday ha spiegato la capacità delle soluzioni di far passare la corrente elettrica grazie alla presenza di ioni in esse, ma lui stesso e altri scienziati, come I. Hittorf e F. Kohlrausch, credevano che gli ioni apparissero sotto l'influenza della corrente. Nel 1884 S. Arrhenius suggerì che in realtà gli ioni si formano semplicemente quando il sale viene sciolto nell'acqua. I lavori di S. Arrhenius, J. van't Hoff e W. Ostwald furono un'importante pietra miliare nello sviluppo della teoria degli elettroliti e delle idee sulle proprietà fisico-chimiche delle soluzioni e sulla loro termodinamica. La corrispondenza tra teoria e dati sperimentali sulla conduttività ionica e sugli equilibri in soluzione divenne più completa dopo che P. Debye ed E. Hückel presero in considerazione le interazioni elettrostatiche a lungo raggio tra ioni nel 1923.
Un importante contributo alla termodinamica elettrochimica e in particolare alla spiegazione della natura del potenziale elettrico (tensione) in una cella elettrochimica e dell'equilibrio tra energia elettrica, chimica e termica è stato dato da J. Gibbs e W. Nernst. Il potenziale elettrochimico è determinato dall'energia chimica dei processi che avvengono nella cellula, ma dipende anche dalla loro velocità (cinetica). La modellazione dei processi cinetici sugli elettrodi è stata effettuata da Y. Tafel (1905), J. Butler (1924), M. Volmer (1930), A. N. Frumkin (1930-1933).
Celle elettrochimiche.
Una cella elettrochimica è solitamente costituita da due semicelle, ciascuna delle quali è un elettrodo immerso nel proprio elettrolita. Gli elettrodi sono realizzati in materiale elettricamente conduttivo (metallo o carbonio) o meno spesso in semiconduttore. I portatori di carica negli elettrodi sono elettroni, mentre i portatori di carica nell'elettrolita sono ioni. Una soluzione acquosa di sale da cucina (cloruro di sodio NaCl), che è un elettrolita, contiene particelle cariche: cationi sodio Na + e anioni cloro Cl –. Se metti una soluzione del genere in un campo elettrico, gli ioni Na+ si sposteranno verso il polo negativo e gli ioni Cl – si sposteranno verso il polo positivo. Anche i sali fusi, come NaCl, sono elettroliti. Gli elettroliti possono essere ad esempio anche solidi B- allumina (polialluminato di sodio) contenente ioni mobili di sodio, o polimeri a scambio ionico.
Le semicelle sono separate da un divisorio che non interferisce con il movimento degli ioni, ma impedisce la miscelazione degli elettroliti. Il ruolo di tale partizione può essere svolto da un ponte salino, un tubo con una soluzione acquosa chiuso su entrambe le estremità con lana di vetro, una membrana a scambio ionico o una lastra di vetro porosa. Entrambi gli elettrodi della cella elettrolitica possono essere immersi nello stesso elettrolita.
Esistono due tipi di celle elettrochimiche: celle galvaniche e celle elettrolitiche (elettrolizzatori). In una cella galvanica, le reazioni chimiche avvengono spontaneamente nell'interfaccia elettrodo/elettrolita e gli elettrodi sono collegati tra loro da un conduttore. Diverse celle galvaniche collegate in serie formano una batteria, una fonte chimica di corrente. In una cella elettrolitica, le reazioni all'interfaccia elettrodo/elettrolita avvengono a causa di una fonte esterna di energia elettrica; quest'ultima viene convertita in energia chimica dai prodotti della reazione che avviene agli elettrodi. La struttura della cella galvanica è mostrata in Fig. 1, e l'elettrolizzatore - in Fig. 2. Si noti che la stessa cella, a seconda della modalità operativa, può comportarsi sia come cella galvanica che come elettrolizzatore. Pertanto, una batteria per auto al piombo agisce come una cella galvanica quando viene utilizzata per avviare il motore (mentre lo scarica) e come un elettrolizzatore quando viene caricata da un generatore o caricabatterie per auto.
Una semplice cella galvanica, creata nel 1836 da J. Daniel (Fig. 1), è costituita da due elettrodi: zinco, immerso in una soluzione acquosa di solfato di zinco, e rame, immerso in una soluzione acquosa di solfato di rame (II). Un tale elemento è simile alle coppie rame-zinco in una colonna voltaica. Quando il circuito esterno è chiuso, gli atomi di zinco presenti sulla superficie dell'elettrodo di zinco si ossidano a ioni con liberazione di elettroni: Zn ® Zn 2+ + 2e – . Questi elettroni si muovono lungo il circuito esterno fino all'elettrodo di rame e riducono gli ioni rame in atomi: Cu 2+ + 2e – ® Cu. Il flusso di elettroni nel circuito esterno è la corrente generata dall'elemento. La reazione complessiva che porta ad una trasformazione chimica e alla generazione di energia elettrica ha la forma
La stessa reazione avviene quando si aggiunge zinco metallico ad una soluzione di solfato di rame, ma in questo caso l'energia chimica viene convertita in energia termica.
Le celle elettrochimiche sono spesso rappresentate schematicamente indicando il confine tra l'elettrodo e l'elettrolita con una barra verticale (| o /) e il ponte salino con due barre (//). Quindi, la cella galvanica in Fig. 1 voce di risposta
dove M è la concentrazione molare della soluzione.
Nella cella elettrolitica mostrata in Fig. 2, si verificano le stesse reazioni degli elettrolizzatori industriali per la produzione di cloro e alcali: la conversione della salamoia (una soluzione acquosa concentrata di cloruro di sodio) in cloro e idrossido di sodio NaOH:
Gli ioni cloruro sull'elettrodo di grafite vengono ossidati in cloro gassoso e l'acqua sull'elettrodo di ferro viene ridotta in ioni idrogeno e idrossido. Gli elettroliti rimangono elettricamente neutri a causa del movimento degli ioni sodio attraverso una partizione: una membrana a scambio ionico. L'elettrodo su cui avviene l'ossidazione (zinco in Fig. 1 e grafite in Fig. 2) è chiamato anodo, e l'elettrodo su cui avviene la riduzione è chiamato catodo.
Potenziale dell'elettrodo.
I principali parametri elettrici delle celle elettrochimiche sono la corrente (misurata in ampere, A) e il potenziale (misurato in volt, V). L'intensità della corrente è determinata dalla velocità delle reazioni degli elettrodi e il potenziale è determinato dall'energia chimica dei processi che si verificano nella cellula. È uguale all'energia (misurata in joule, J) divisa per la quantità di elettricità (misurata in coulomb, C), cioè 1 V = 1 J / Cl. Pertanto, il potenziale di un elemento (forza elettromotrice, fem) è una misura dell'energia generata durante le reazioni che si verificano in esso. Se il circuito esterno è aperto, non si verificano reazioni dell'elettrodo.
Il potenziale di una cella galvanica con circuito esterno aperto fornisce informazioni sulla termodinamica delle sue reazioni. Il potenziale dell'elemento mostrato in Fig. 1, a concentrazioni di soluzione di 1 M e una temperatura di 25 ° C - il suo potenziale standard E° – pari a 1,10 V. Energia corrispondente, potenziale termodinamico di Gibbs, D G°, è dato da
Dove N– il numero di elettroni trasferiti durante la reazione (in questo caso 2), F– Numero di Faraday (96 485 C / neo). Il potenziale di una cella galvanica è pari alla differenza di potenziale tra le sue due semicelle, cioè la differenza tra i suoi potenziali di elettrodo. I potenziali degli elettrodi vengono misurati rispetto al potenziale dell'elettrodo di riferimento, che è convenzionalmente considerato pari a zero ( cm. tavolo). Di comune accordo, come elettrodo standard è stato selezionato un normale elettrodo a idrogeno (N.H.E.); si tratta di una lastra di platino, rivestita di nero di platino, saturata con gas idrogeno alla pressione di 1,01H 10 5 Pa (1 atm.) e immersa in una soluzione contenente ioni H+ ad attività termodinamica UN= 1. Schematicamente questo elettrodo può essere rappresentato come Pt/H 2 (1.01H 10 5 Pa)/H + ( UN= 1). Su di esso avviene la reazione 2H + + 2e – ® H 2. Per determinare il potenziale standard di un elettrodo di rame Cu/Cu 2+, assemblare la seguente cella galvanica:
e per la semireazione Cu 2+ + 2e – ® Cu la misurazione dà
Analogamente, per la semicella Zn/Zn 2+, in cui avviene la reazione Zn 2+ + 2e – ® Zn, otteniamo
La differenza tra questi due potenziali degli elettrodi standard è uguale al potenziale standard dell'elemento Zn-Cu.
L'elettrodo a idrogeno, infatti, viene utilizzato raramente nelle misure potenziometriche, poiché rappresenta un sistema ideale di difficile implementazione nella pratica. Molto più spesso vengono utilizzati elettrodi di confronto più convenienti e compatti di vario tipo, aventi un valore potenziale specifico e attentamente misurato rispetto alla corrente. Di solito utilizzano un elettrodo al calomelano (CE), costituito da mercurio metallico, cloruro di mercurio (calomelano) e una soluzione di cloruro di potassio: Hg /Hg 2 Cl 2 /KCl. All'elettrodo avviene la seguente reazione:
Potenziale k.e. dipende dalla concentrazione degli ioni mercurio e quest'ultimo dipende dalla concentrazione della soluzione KCl. Per soluzione satura di KCl E° (k.e.) av = 0,2412 V (n.e.) a 25° C.
|
POTENZIALI DEGLI ELETTRODI STANDARD |
|
| Reazione dell'elettrodo |
E o, V |
| Li + + e – ® Li | |
| Mg2+ + 2e – ® Mg | |
| Al 3+ + 3e – ® Al | |
| Zn 2+ + 2e – ® Zn | |
| Cr 3+ + e – ® Cr 2+ | |
| 2H + + 2e – ® H 2 | |
| Cu 2+ + 2e – ® Cu | |
| Fe 3+ + e – ® Fe 2+ | |
| O2+4H++4e – ®2H2O | |
| Cl2 + 2e – ® 2Cl – | |
| F 2 + 2e – ® 2F – | |
Si noti che la sostanza di alcuni elettrodi non è inclusa nell'equazione della reazione corrispondente. Sì, reazione
avviene effettivamente su un elettrodo di platino in una cella Pt/Fe 3+, Fe 2+. L'elettrodo di platino è inerte e fornisce contatto solo con l'elettrolita contenente le forme ossidate e ridotte dell'elemento (in questo caso ioni ferro bivalenti e trivalenti). L'elettrodo di platino svolge lo stesso ruolo in N.V.E.
Le tabelle dei potenziali degli elettrodi consentono di calcolare la FEM di una cella galvanica in base ai potenziali degli elettrodi. Possono anche essere utilizzati per prevedere se si verificherà una particolare reazione redox. Possiamo parlare di potenziale di elettrodo standard solo quando l'attività dei componenti che partecipano alla reazione è uguale a 1, cioè la loro concentrazione in soluzione è vicina a 1M. Potenziale dell'elettrodo E dipende dalla concentrazione delle forme ossidate e ridotte in soluzione ed è correlato ad esse e al potenziale standard E° Equazione di Nernst. Per una reazione generalizzata
bue + N e – = rosso
questa equazione ha la forma
Dove R– costante universale dei gas, T– temperatura assoluta, e – attività delle forme ossidate e ridotte. Le attività dei solidi e dei liquidi puri sono considerate 1. A 25° C RT/F= 0,025 V. Misurando i potenziali dell'elettrodo rispetto al potenziale dell'elettrodo di riferimento, è possibile determinare le concentrazioni di sostanze nella soluzione utilizzando l'equazione (10); questo metodo è chiamato potenziometria.
Reazioni degli elettrodi.
Le misurazioni potenziometriche vengono eseguite in condizioni in cui non c'è corrente nella cella elettrochimica. Ciò significa che in esso non si verificano cambiamenti chimici complessivi e il potenziale misurato (equilibrio) è determinato dalla termodinamica delle reazioni. In queste condizioni fattori come la dimensione e la forma degli elettrodi o l'intensità dell'agitazione della soluzione non influenzano il potenziale misurato. Se la corrente scorre attraverso una cella elettrochimica, la velocità delle reazioni degli elettrodi dipende non solo dai parametri termodinamici, ma anche dall'intensità della corrente secondo l'equazione
Dove N– numero di elettroni che partecipano a una data reazione elettrodica, F– Numero di Faraday. In questo caso, il potenziale della cella elettrochimica dipende da fattori cinetici, nonché dal materiale di cui è composto l'elettrodo, dalle dimensioni e dalla forma dell'elettrodo, dall'intensità dell'agitazione della soluzione e da molti altri fattori. La resistenza interna della cella non può essere trascurata. Oltre alla differenza di potenziale ai confini di entrambi gli elettrodi/elettroliti, nella soluzione stessa si verifica una caduta di tensione, a causa della sua resistenza. Questa caduta di tensione rende difficile studiare gli effetti associati alle reazioni che si verificano su entrambi gli elettrodi. Di solito, la reazione viene studiata su un elettrodo, chiamato elettrodo di lavoro o indicatore, utilizzando una cella a tre elettrodi (Fig. 3): il terzo elettrodo (ad esempio, calomelano saturo) è posizionato nello stesso compartimento dell'elettrodo di lavoro uno, il più vicino possibile ad esso per ridurre al minimo l'effetto della caduta di tensione ohmica. Misurando la corrente attraverso l'elettrodo di lavoro in funzione del potenziale di questo elettrodo rispetto all'elettrodo di riferimento, il cosiddetto curva di polarizzazione.
Quando viene fatta passare una corrente esterna, il potenziale dell'elettrodo differisce da quello di equilibrio. Questa deviazione è chiamata polarizzazione e la sua grandezza è chiamata sovratensione. La sovratensione dipende da diversi fattori che limitano la velocità delle reazioni degli elettrodi. Le reazioni veloci degli elettrodi a una data densità di corrente (intensità di corrente per unità di superficie dell'elettrodo) si verificano a potenziali vicini a quelli termodinamici e quindi a bassa sovratensione. Le reazioni lente sono caratterizzate da un'elevata sovratensione. La velocità delle reazioni degli elettrodi, e quindi la sovratensione, dipende dalla concentrazione dei reagenti, dalla temperatura, dal solvente, dal materiale dell'elettrodo, dal metodo e dalla velocità di trasferimento di massa e dalla densità di corrente. La sovratensione totale può essere scomposta in diverse componenti: concentrazione, attivazione e reazione.
La sovratensione di concentrazione è causata dal fatto che quando passa una corrente, la concentrazione dello ione reagente sulla superficie dell'elettrodo cambia, poiché in quest'area si consumano sostanze elettroattive e si formano prodotti di reazione. Consideriamo la riduzione di Cu 2+ su un elettrodo di rame. Inizialmente, la concentrazione di Cu 2+ nella soluzione è 1M. In assenza di corrente nel circuito, il potenziale dell'elettrodo di rame è vicino al potenziale standard della coppia Cu/Cu 2+, cioè 0,34 V rispetto a N.E. [ cm. equazione (6)]. Al passaggio della corrente catodica, la concentrazione di ioni Cu 2+ sulla superficie dell'elettrodo diminuisce e il potenziale catodico, secondo l'equazione di Nernst (10), diventa sempre più negativo. Porzioni fresche del reagente arrivano dalla soluzione all'elettrodo in diversi modi: come risultato di diffusione, convezione e migrazione. Maggiore è la velocità di questi processi (ad esempio, più intensa è la miscelazione), minore è la sovratensione di concentrazione. Questa componente della sovratensione totale può spesso essere calcolata. Se la polarizzazione della concentrazione fornisce il contributo principale alla sovratensione totale (ciò significa che la velocità delle restanti fasi della reazione dell'elettrodo è elevata), la reazione viene chiamata reversibile o Nernstiana.
La sovratensione di attivazione si verifica a causa del fatto che il trasferimento di elettroni sulla superficie dell'elettrodo non avviene istantaneamente, ma a una velocità finita. Consideriamo la reazione generalizzata dell'elettrodo ox + N e – = rosso. Per trasferire gli elettroni ai composti ossidati ad una data velocità (cioè ad una data densità di corrente), è necessario superare una barriera energetica chiamata energia di attivazione della reazione dell'elettrodo. Questa energia è fornita dal potenziale applicato. La relazione tra densità di corrente e sovratensione di attivazione è descritta dalle equazioni di Butler-Volmer e Tafel, che possono essere utilizzate per determinare i parametri cinetici delle reazioni degli elettrodi. Molte reazioni degli elettrodi, come la riduzione dell'acqua in idrogeno e la sua ossidazione in ossigeno, avvengono lentamente. La velocità di reazione dell'elettrodo può dipendere in gran parte dal materiale di cui è costituito l'elettrodo e dalle proprietà della sua superficie. Ad esempio, su un elettrodo di mercurio, la riduzione dell'acqua in idrogeno è notevolmente difficile: è caratterizzata da un'elevata sovratensione dell'idrogeno. Questa reazione avviene molto più velocemente e con meno sovratensione sul platino; È per questo motivo che nell'elettrodo di riferimento dell'idrogeno viene utilizzato il platino. La velocità delle reazioni dipende anche dalle sostanze adsorbite o legate alla superficie dell'elettrodo; Pertanto, lo ione cianuro e numerosi composti organici riducono la velocità di sviluppo dell'idrogeno sulla superficie dell'elettrodo di platino. Allo stesso tempo, alcuni tensioattivi possono aumentare significativamente la velocità di reazione degli elettrodi: sono chiamati elettrocatalizzatori.
La sovratensione di reazione si verifica quando il trasferimento di elettroni sull'elettrodo è accoppiato con una reazione chimica nella soluzione. Una tale reazione può fungere da fonte di particelle coinvolte nel trasferimento di elettroni e allo stesso tempo limitare la velocità dell'intero processo dell'elettrodo. Questo è il motivo per cui è così importante conoscere i dettagli del meccanismo (cioè stadi e stati intermedi) delle reazioni degli elettrodi. In molti casi, il materiale di partenza subisce diverse trasformazioni prima di diventare il prodotto finale all'elettrodo, come nel caso della riduzione dell'ossigeno in acqua, processo di grande importanza pratica. La reazione totale ha la forma
e consiste di diverse fasi, in una delle quali il legame ossigeno-ossigeno viene rotto. A causa di questa reazione a più stadi, la reazione sulla maggior parte degli elettrodi è lenta e su scala industriale viene effettuata in presenza di elettrocatalizzatori. Il meccanismo delle reazioni degli elettrodi viene studiato utilizzando i metodi elettroanalitici descritti di seguito. Spesso il corso di una reazione cambia quando cambiano la composizione della soluzione e la natura del solvente. Ad esempio, la riduzione dell'ossigeno nell'acetonitrile, dove c'è carenza di protoni, procede secondo un semplice meccanismo ad un elettrone:
Metodi di analisi elettrochimica.
Sono stati sviluppati diversi metodi elettrochimici per l'analisi qualitativa e quantitativa delle sostanze chimiche, spesso utili anche per determinare i parametri termodinamici e cinetici delle reazioni elettrodiche e studiarne i meccanismi.
Conduttometria
si basa sulla misurazione della conduttività elettrica di una soluzione e viene utilizzato per determinare la concentrazione di sali, acidi, basi, ecc. Nelle determinazioni conduttometriche vengono solitamente utilizzati elettrodi costituiti da materiali identici e le condizioni per la loro condotta sono selezionate in modo tale da ridurre al minimo il contributo dei salti di potenziale su entrambe le interfacce elettrodo/elettrolita (ad esempio, viene utilizzata corrente alternata ad alta frequenza ). In questo caso il contributo principale al potenziale di cella misurato è dato dalla caduta di tensione ohmica IR, Dove R– resistenza della soluzione. La conduttività elettrica di una soluzione monocomponente può essere correlata alla sua concentrazione e la misurazione della conduttività elettrica di elettroliti di composizione complessa consente di stimare il contenuto totale di ioni nella soluzione e viene utilizzata, ad esempio, per monitorare la qualità del distillato o acqua deionizzata. In un altro tipo di conduttometria - la titolazione conduttimetrica - un reagente noto viene aggiunto in porzioni alla soluzione analizzata e viene monitorata la variazione della conduttività elettrica. Il punto di equivalenza, in cui si nota un brusco cambiamento nella conduttività elettrica, è determinato da un grafico della dipendenza di questo valore dal volume del reagente aggiunto.
Potenziometria
utilizzato per determinare vari parametri fisico-chimici basati sui dati sul potenziale di una cella galvanica. Il potenziale dell'elettrodo in assenza di corrente nel circuito elettrochimico, misurato rispetto all'elettrodo di riferimento, è correlato alla concentrazione della soluzione mediante l'equazione di Nernst. Nelle misurazioni potenziometriche vengono ampiamente utilizzati elettrodi ionoselettivi sensibili principalmente a uno ione in soluzione: un elettrodo di vetro per la misurazione del pH ed elettrodi per la determinazione selettiva di ioni sodio, ammonio, fluoro, calcio, magnesio, ecc. Lo strato dell'elettrodo ionoselettivo può includere enzimi e il risultato è un sistema sensibile al substrato appropriato. Si noti che il potenziale di un elettrodo ionoselettivo non è determinato dal trasferimento di elettroni, come nel caso di sostanze con conduttività elettronica, ma principalmente dal trasferimento o dallo scambio di ioni. Tuttavia, l'equazione di Nernst, che mette in relazione il potenziale dell'elettrodo con il logaritmo della concentrazione (o attività) di una sostanza in soluzione, è applicabile anche a tale elettrodo. Nella titolazione potenziometrica, il reagente viene aggiunto in porzioni alla soluzione da analizzare e viene monitorata la variazione di potenziale. Le curve a forma di S caratteristiche di questo tipo di titolazione permettono di determinare il punto di equivalenza e trovare parametri termodinamici come la costante di equilibrio e il potenziale standard.
Voltammetria.
Tutte le varietà di metodi voltammetrici utilizzano un microelettrodo funzionante con una superficie di 10–7–10–1 cm2. Le curve corrente-tensione ottenute con il suo aiuto consentono di identificare le sostanze disciolte, determinarne la concentrazione e spesso i parametri termodinamici e cinetici. Il primo metodo voltammetrico - la polarografia - fu proposto nel 1922 dal chimico ceco J. Heyrovsky. L'elettrodo funzionante nella sua configurazione era un elettrodo di mercurio gocciolante. Il mercurio ha un'elevata sovratensione dell'idrogeno, quindi un elettrodo di mercurio è conveniente per studiare i processi che si verificano a potenziali negativi. La superficie dell'elettrodo viene costantemente rinnovata durante il processo di misurazione, eliminando così la contaminazione dell'elettrodo. Gli studi voltammetrici vengono eseguiti anche utilizzando elettrodi solidi, come platino e carbonio, e utilizzano processi che si verificano a potenziali positivi. Nella voltammetria a potenziale lineare (cronoamperometria), il potenziale cambia linearmente con il tempo e la soluzione non viene agitata, quindi il trasferimento di massa avviene esclusivamente per diffusione. Nella voltammetria ciclica, impulsi di tensione triangolari ripetuti vengono applicati a un elettrodo. Le sostanze formate nella sezione ascendente del ciclo vengono studiate nella sua sezione discendente. Questo metodo è particolarmente efficace per studiare il meccanismo delle reazioni degli elettrodi analizzando le curve di polarizzazione a diverse velocità di scansione potenziali e diverse concentrazioni di soluzione. Esistono altri tipi di voltammetria - impulso differenziale e onda quadra - in cui impulsi di tensione di forme diverse si sovrappongono a un potenziale crescente linearmente. Questi metodi sono ampiamente utilizzati per determinare piccole concentrazioni di sostanze in soluzione. Se durante una misurazione voltammetrica la soluzione viene miscelata, il che significa che il trasferimento di massa avviene contemporaneamente per convezione e diffusione, allora si parla di voltammetria idrodinamica. In questo caso è conveniente utilizzare un elettrodo a disco rotante, in quanto le curve sperimentali corrente-tensione possono essere direttamente confrontate con quelle teoriche.
Amperometria.
Il metodo si basa sulla misurazione della corrente di diffusione limite che passa attraverso una soluzione a tensione fissa tra l'elettrodo indicatore e l'elettrodo di riferimento. Nella titolazione amperometrica, il punto equivalente è determinato dall'interruzione della curva corrente - il volume della soluzione di lavoro aggiunta. I metodi cronoamperometrici si basano sulla misurazione della dipendenza della corrente dal tempo e vengono utilizzati principalmente per determinare i coefficienti di diffusione e le costanti di velocità. Le celle elettrochimiche in miniatura che fungono da sensori all'uscita delle colonne del cromatografo liquido funzionano secondo il principio dell'amperometria (così come della voltammetria). I metodi galvanostatici sono simili a quelli amperometrici, ma misurano il potenziale quando una certa quantità di corrente passa attraverso la cella. Pertanto, nella cronopotenziometria, viene controllata la variazione del potenziale nel tempo. Questi metodi vengono utilizzati principalmente per studiare la cinetica delle reazioni degli elettrodi.
Coulometria.
Nella coulometria, a un potenziale controllato, l'elettrolisi completa di una soluzione viene effettuata miscelandola intensamente in un elettrolizzatore con un elettrodo di lavoro relativamente grande (rete di mercurio o platino inferiore). Quantità totale di elettricità ( Q, C) richiesta per l'elettrolisi è correlata alla quantità di sostanza in formazione ( UN, d) Legge di Faraday:
Dove M- dicono massa (g/mol), F-Numero di Faraday. La titolazione coulometrica prevede l'utilizzo di una corrente costante per generare elettroliticamente un reagente che reagisce con la sostanza da determinare. L'avanzamento della titolazione è controllato potenziometricamente o amperometricamente. I metodi coulometrici sono convenienti perché sono di natura assoluta (ovvero consentono di calcolare la quantità dell'analita senza ricorrere a curve di calibrazione) e sono insensibili ai cambiamenti nelle condizioni di elettrolisi e nei parametri dell'elettrolizzatore (area superficiale dell'elettrodo o intensità di agitazione). Nella coulogravimetria, la quantità di sostanza che ha subito l'elettrolisi viene determinata pesando l'elettrodo prima e dopo l'elettrolisi.
Esistono altri metodi elettroanalitici. Nella polarografia a corrente alternata, una tensione sinusoidale di bassa ampiezza viene applicata a un potenziale che varia linearmente su un ampio intervallo di frequenze e vengono determinate l'ampiezza e lo sfasamento della corrente alternata risultante o l'impedenza. Da questi dati si ottengono informazioni sulla natura delle sostanze in soluzione e sul meccanismo e la cinetica delle reazioni degli elettrodi. I metodi a strato sottile utilizzano celle elettrochimiche con uno strato di elettrolita spesso 10–100 µm. In tali celle, l'elettrolisi procede più velocemente rispetto agli elettrolizzatori convenzionali. Per studiare i processi degli elettrodi vengono utilizzati metodi spettrochimici con registrazione spettrofotometrica. Per analizzare le sostanze formate sulla superficie dell'elettrodo, viene misurato il loro assorbimento della luce nelle regioni visibile, UV e IR. I cambiamenti nelle proprietà della superficie dell'elettrodo e del mezzo vengono monitorati utilizzando metodi di riflessione elettrica ed ellissometria, che si basano sulla misurazione della riflessione della radiazione dalla superficie dell'elettrodo. Questi includono metodi di riflessione speculare e diffusione Raman della luce (spettroscopia Raman), spettroscopia di seconda armonica (spettroscopia di Fourier).
Altri fenomeni e metodi elettrochimici.
Con il movimento relativo dell'elettrolita e delle particelle o superfici cariche, si verificano effetti elettrocinetici. Un esempio importante di questo tipo è l'elettroforesi, in cui avviene la separazione di particelle cariche (ad esempio molecole proteiche o particelle colloidali) che si muovono in un campo elettrico. I metodi elettroforetici sono ampiamente utilizzati per separare proteine o acidi desossiribonucleici (DNA) nei gel. I fenomeni elettrici svolgono un ruolo importante nel funzionamento degli organismi viventi: sono responsabili della generazione e della propagazione degli impulsi nervosi, della comparsa di potenziali transmembrana, ecc. Vari metodi elettrochimici vengono utilizzati per studiare i sistemi biologici e i loro componenti. È interessante anche studiare l'effetto della luce sui processi elettrochimici. Pertanto, l'oggetto della ricerca fotoelettrochimica è la generazione di energia elettrica e l'avvio di reazioni chimiche sotto l'influenza della luce, che è molto importante per aumentare l'efficienza della conversione dell'energia solare in energia elettrica. Qui vengono comunemente utilizzati elettrodi semiconduttori realizzati in biossido di titanio, solfuro di cadmio, arseniuro di gallio e silicio. Un altro fenomeno interessante è l’elettrochemiluminescenza, cioè generazione di luce in una cella elettrochimica. Si osserva quando sugli elettrodi si formano prodotti ad alta energia. Spesso il processo viene eseguito in modo ciclico per ottenere sia la forma ossidata che quella ridotta di un dato composto. La loro interazione tra loro porta alla formazione di molecole eccitate, che passano allo stato fondamentale con l'emissione di luce.
Elettrochimica applicata.
L'elettrochimica ha molte applicazioni pratiche. Con l'ausilio di celle galvaniche primarie (elementi usa e getta) collegate alle batterie, l'energia chimica viene convertita in energia elettrica. Fonti di corrente secondarie - batterie - immagazzinano energia elettrica. Le celle a combustibile sono fonti di energia primarie che generano elettricità attraverso una fornitura continua di reagenti (come idrogeno e ossigeno). Questi principi sono alla base delle fonti di energia portatili e delle batterie utilizzate nelle stazioni spaziali, nei veicoli elettrici e nei dispositivi elettronici.
La produzione su larga scala di molte sostanze si basa sulla sintesi elettrochimica. L'elettrolisi della salamoia nel processo cloro-alcali produce cloro e alcali, che vengono poi utilizzati per produrre composti organici e polimeri, nonché nell'industria della pasta e della carta. I prodotti dell'elettrolisi sono composti come clorato di sodio, persolfato, permanganato di sodio; I metalli industrialmente importanti si ottengono mediante elettroestrazione: alluminio, magnesio, litio, sodio e titanio. È preferibile utilizzare sali fusi come elettroliti, poiché in questo caso, a differenza delle soluzioni acquose, la riduzione dei metalli non è complicata dal rilascio di idrogeno. Il fluoro è prodotto mediante elettrolisi in sale fuso. I processi elettrochimici servono come base per la sintesi di alcuni composti organici; ad esempio, l'adiponitrile (un intermedio nella sintesi del nylon) si ottiene mediante idrodimerizzazione dell'acrilonitrile.
La galvanica di argento, oro, cromo, ottone, bronzo e altri metalli e leghe è ampiamente praticata su vari oggetti per proteggere i prodotti in acciaio dalla corrosione, per scopi decorativi, per la fabbricazione di connettori elettrici e circuiti stampati nell'industria elettronica. I metodi elettrochimici vengono utilizzati per la lavorazione dimensionale ad alta precisione di pezzi in metalli e leghe, in particolare quelli che non possono essere lavorati con metodi meccanici convenzionali, nonché per la produzione di parti con profili complessi. Quando la superficie di metalli come alluminio e titanio viene anodizzata, si formano pellicole protettive di ossido. Tali pellicole vengono create sulla superficie di pezzi in alluminio, tantalio e niobio nella produzione di condensatori elettrolitici e talvolta per scopi decorativi.
I metodi elettrochimici vengono spesso utilizzati come base per gli studi sui processi di corrosione e la selezione dei materiali che rallentano questi processi. La corrosione delle strutture metalliche può essere prevenuta utilizzando la protezione catodica, per la quale una fonte esterna è collegata alla struttura da proteggere e l'anodo e la struttura vengono mantenuti ad un potenziale tale da escluderne l'ossidazione. Sono in fase di studio le possibilità di applicazione pratica di altri processi elettrochimici. Quindi, l'elettrolisi può essere utilizzata per purificare l'acqua. Una direzione molto promettente è la conversione dell'energia solare utilizzando metodi fotochimici. Sono in fase di sviluppo monitor elettrochimici, il cui principio di funzionamento si basa sull'elettrochemiluminescenza. Menzioniamo anche lo studio dei cambiamenti reversibili nel colore della superficie dell'elettrodo a seguito delle reazioni degli elettrodi.
Letteratura:
Agladze R.I. Elettrochimica applicata. M.-L., 1975
Izmailov N.A. Elettrochimica delle soluzioni. M., 1976
Metodi di misura in elettrochimica, vol. 1–2. M., 1977
Koryta I. Ioni, elettrodi, membrane. M., 1983
Bagotsky V.S. Nozioni di base di elettrochimica. M., 1988