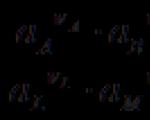เคมีไฟฟ้า. เซลล์กัลวานิก เซลล์เคมีไฟฟ้า
องค์ประกอบไฟฟ้าเคมี แรงเคลื่อนไฟฟ้า อุณหพลศาสตร์ของเซลล์กัลวานิก การวัดแรงเคลื่อนไฟฟ้า
ไฟฟ้าสองชั้น กลไกการเกิดและโครงสร้าง
ธาตุกัลวานิก แรงเคลื่อนไฟฟ้า.
เมื่อกระแสไฟฟ้าไหลผ่านอิเล็กโทรไลต์ ปฏิกิริยาเคมีไฟฟ้าจะเกิดขึ้นบนพื้นผิวของอิเล็กโทรด การเกิดปฏิกิริยาเคมีไฟฟ้าสามารถเกิดขึ้นได้จากแหล่งกระแสภายนอก ปรากฏการณ์ตรงกันข้ามก็เป็นไปได้เช่นกัน: ปฏิกิริยาเคมีไฟฟ้าที่เกิดขึ้นบนอิเล็กโทรดสองตัวที่แช่อยู่ในอิเล็กโทรไลต์จะทำให้เกิดกระแสไฟฟ้า และปฏิกิริยาจะเกิดขึ้นในวงจรปิดเท่านั้น (เมื่อกระแสไหลผ่าน)
เซลล์ไฟฟ้าเคมี (หรือกัลวานิก) เป็นอุปกรณ์สำหรับผลิตกระแสไฟฟ้าผ่านปฏิกิริยาเคมีไฟฟ้า องค์ประกอบไฟฟ้าเคมีที่ง่ายที่สุดประกอบด้วยอิเล็กโทรดโลหะสองตัว (ตัวนำชนิดแรก) ลดลงเป็นอิเล็กโทรไลต์ (ตัวนำชนิดที่สอง) และเชื่อมต่อกันด้วยหน้าสัมผัสโลหะ ธาตุไฟฟ้าเคมีหลายชนิดเชื่อมต่อกันในรูปแบบอนุกรม วงจรไฟฟ้าเคมี .
ลักษณะเชิงปริมาณที่สำคัญที่สุดขององค์ประกอบไฟฟ้าเคมีคือแรงเคลื่อนไฟฟ้า(EMF,E) ซึ่งเท่ากับค่าความต่างศักย์ เปิดองค์ประกอบอย่างถูกต้อง (อันหนึ่งซึ่งตัวนำชนิดที่หนึ่งจากวัสดุเดียวกันเชื่อมต่อกับอิเล็กโทรดสุดท้ายขององค์ประกอบ)
หากเมื่อกระแสไฟฟ้าไหลไปในทิศทางที่แตกต่างกันปฏิกิริยาเดียวกันเกิดขึ้นบนพื้นผิวของอิเล็กโทรด แต่ไปในทิศทางตรงกันข้ามอิเล็กโทรดดังกล่าวรวมถึงองค์ประกอบหรือวงจรที่ประกอบด้วยพวกมันจะถูกเรียกว่า ย้อนกลับได้ . แรงเคลื่อนไฟฟ้าขององค์ประกอบที่พลิกกลับได้คือคุณสมบัติทางอุณหพลศาสตร์เช่น ขึ้นอยู่กับ T, P ลักษณะของสารที่ประกอบเป็นอิเล็กโทรดและสารละลาย และความเข้มข้นของสารละลายเหล่านี้เท่านั้น ตัวอย่างขององค์ประกอบที่พลิกกลับได้คือ องค์ประกอบของแดเนียล-จาโคบี :
(-) Cu çZn çZnSO 4 ççCuSO 4 çCu (+)
ซึ่งแต่ละอิเล็กโทรดสามารถย้อนกลับได้ เมื่อองค์ประกอบทำงาน จะเกิดปฏิกิริยาต่อไปนี้: Zn ® Zn 2+ + 2 จ, Cu 2+ + 2 จ® ลูกบาศ์ก เมื่อกระแสที่มีความแรงน้อยถูกส่งผ่านจากแหล่งภายนอก จะเกิดปฏิกิริยาย้อนกลับบนอิเล็กโทรด
ตัวอย่างขององค์ประกอบที่ไม่สามารถย้อนกลับได้คือ ธาตุโวลต้า :
(-) สังกะสี ç H 2 SO 4 ç Cu (+)
เมื่อองค์ประกอบทำงาน จะเกิดปฏิกิริยาต่อไปนี้: Zn ® Zn 2+ + 2 จ, 2H + + 2 จ® เอช 2 . เมื่อส่งกระแสจากแหล่งภายนอก ปฏิกิริยาอิเล็กโทรดจะเป็น: 2H + + 2 จ® H 2 , Cu ® Cu 2+ + 2 จ .
EMF ขององค์ประกอบไฟฟ้าเคมีเป็นค่าบวก เนื่องจาก มันสอดคล้องกับกระบวนการที่เกิดขึ้นเองซึ่งก่อให้เกิดผลงานเชิงบวก กระบวนการย้อนกลับซึ่งไม่สามารถเกิดขึ้นได้โดยอิสระจะสอดคล้องกับ EMF เชิงลบ เมื่อสร้างห่วงโซ่ขององค์ประกอบไฟฟ้าเคมี กระบวนการในองค์ประกอบหนึ่งสามารถถูกกำกับเพื่อให้มาพร้อมกับค่าใช้จ่ายของงานจากภายนอก (กระบวนการที่ไม่เกิดขึ้นเอง) โดยใช้สำหรับการทำงานขององค์ประกอบอื่นของห่วงโซ่ใน ซึ่งเป็นกระบวนการที่เกิดขึ้นเอง แรงเคลื่อนไฟฟ้ารวมของวงจรใดๆ เท่ากับผลรวมพีชคณิตของปริมาณบวกและลบ ดังนั้นจึงเป็นสิ่งสำคัญมากในการเขียนแผนภาพวงจรโดยคำนึงถึงสัญญาณของ EMF โดยใช้กฎที่ยอมรับ
แรงเคลื่อนไฟฟ้าของวงจรไฟฟ้าเคมีถือเป็นบวกหากเมื่อเขียนวงจร อิเล็กโทรดด้านขวาจะมีประจุบวกสัมพันธ์กับด้านซ้าย (ในระหว่างการทำงานของวงจร แคตไอออนจะผ่านสารละลายจากอิเล็กโทรดที่เขียนทางด้านซ้ายไปยังอิเล็กโทรดที่เขียนทางด้านขวา และอิเล็กตรอนเคลื่อนที่เข้า ทิศทางเดียวกันในวงจรภายนอก) ตัวอย่าง.
อุณหพลศาสตร์ของเซลล์กัลวานิก .
ปล่อยให้ปฏิกิริยาดำเนินไปแบบย้อนกลับและมีอุณหภูมิคงที่ในระบบไฟฟ้าเคมี:
n A A + n B B + ... ± nF Û n L L + n M M + ... ±
พลังงานไฟฟ้าที่สร้างโดยองค์ประกอบนั้นเท่ากับงานที่มีประโยชน์ A¢ ของกระบวนการทั้งหมด งานที่มีประโยชน์ A¢ ของกระบวนการแบบพลิกกลับได้มีค่าสูงสุด และที่ P,T = const เท่ากับการลดลงของศักยภาพไอโซบาริกของระบบ:
DG P, T = nFE P, T
E P , T - EMF แบบย้อนกลับของระบบ
อี P,T = -DG P,T / nF , EV,T = -DF V,T / nF
ดังนั้นด้วยการวัด EMF ขององค์ประกอบและค่าสัมประสิทธิ์อุณหภูมิ จึงเป็นไปได้ที่จะค้นหาค่าของ DG และ DS สำหรับกระบวนการทั้งหมดที่เกิดขึ้นในเซลล์กัลวานิก กระบวนการนี้เกิดขึ้นเอง ดังนั้น DG< 0.
เมื่อใช้สมการ Gibbs-Helmholtz เราสามารถคำนวณการเปลี่ยนแปลงเอนทาลปีของกระบวนการได้:
DH = DG - T = -nFE P + TnF
nFE P = -DH + nFT = + nFT
nFE V = -DU + nFT = + nFT
จากสมการพบว่าความสัมพันธ์ระหว่างพลังงานไฟฟ้าที่สร้างขึ้นหรือดูดซับในระบบไฟฟ้าเคมีแบบย้อนกลับกับผลกระทบทางความร้อนของปฏิกิริยาที่เกิดขึ้นนั้นขึ้นอยู่กับเครื่องหมายและขนาดของค่าสัมประสิทธิ์อุณหภูมิของแรงเคลื่อนไฟฟ้า ดีอี/ ดีที :
1. ถ้าดีอี / ดีที > 0 จากนั้น nFE > (DG > DH) และระบบจะแปลงเป็นพลังงานไฟฟ้า ไม่เพียงแต่ปริมาณความร้อนที่สอดคล้องกับผลกระทบทางความร้อนของปฏิกิริยา แต่ยังรวมถึงความร้อนเพิ่มเติมด้วย - ความอบอุ่นของเปเลเทียร์ Q P = nFT ดีอี/ ดีทียืมมาจากสิ่งแวดล้อม ภายใต้สภาวะอะเดียแบติก (ภายใต้เงื่อนไขของฉนวนกันความร้อน เมื่อไม่สามารถแลกเปลี่ยนกับสิ่งแวดล้อมได้) ค่า T ของระบบจะลดลง การระบายความร้อนของระบบจะสังเกตเห็นได้ชัดเจนเป็นพิเศษหาก ดีอี/ ดีที > 0 < 0 (реакция эндотермична).
2. ถ้าดีอี / ดีที < 0 จากนั้น กฟผ< (DG < DH) и часть теплоты реакции будет рассеиваться в виде теплоты Пелетье. В адиабатическом режиме система будет нагреваться.
3. ถ้าดีอี / ดีที = 0 จากนั้น DG = DH และ nFE = - พลังงานไฟฟ้าที่ผลิตแบบผันกลับได้โดยระบบจะเทียบเท่ากับผลกระทบทางความร้อนของปฏิกิริยาเคมี ความสัมพันธ์นี้เรียกว่า หลักการของทอมสัน (กฎ) .
ในการคำนวณ EMF สามารถเขียนสมการใหม่ได้เป็น:
เมื่อใช้สมการก็ต้องจำไว้ว่า ใช้ได้กับระบบไฟฟ้าเคมีแบบผันกลับได้เท่านั้นดังนั้นเมื่อศึกษาการพึ่งพา EMF กับ T จำเป็นต้องหลีกเลี่ยงการใช้ระบบไฟฟ้าเคมีที่มีขอบเขตของของเหลวเพราะ ศักยภาพการแพร่กระจายที่เกิดขึ้นไม่สมดุล
ให้เราเชื่อมโยง EMF ขององค์ประกอบกับค่าคงที่สมดุลของปฏิกิริยาที่เกิดขึ้นในองค์ประกอบ สมการไอโซเทอมของปฏิกิริยาเคมี:
DG = RT lnเค ก - RT
อี = - = lnเค ก -
เทอมแรกทางด้านขวาของสมการสำหรับ P, T ที่กำหนดคือค่าคงที่ สามารถเขียนแทนด้วย E o อี โอ - EMF มาตรฐานขององค์ประกอบ (ระบบไฟฟ้าเคมี) , เช่น. EMF เลย ฉัน = 0.
อี = อี โอ + ln
 = อี โอ + 2.303 แอลจี
= อี โอ + 2.303 แอลจี
ดังนั้น EMF ของระบบไฟฟ้าเคมีจึงเป็นหน้าที่ของกิจกรรมของผู้เข้าร่วมในปฏิกิริยาเคมีไฟฟ้า สมการข้างต้นทำให้สามารถคำนวณค่าของ DG และ K ได้ กขึ้นอยู่กับค่าทดลองของ E และในทางกลับกันคำนวณ E โดยรู้ลักษณะทางอุณหพลศาสตร์ของปฏิกิริยาเคมี
การวัดแรงเคลื่อนไฟฟ้า .
ในการวัดค่าสมดุล (ย้อนกลับได้) ของ EMF ขององค์ประกอบไฟฟ้าเคมี จำเป็นที่กระบวนการจะเกิดขึ้นอย่างช้าๆ อย่างไม่มีที่สิ้นสุด เช่น เพื่อให้องค์ประกอบทำงานที่กระแสไฟฟ้าเพียงเล็กน้อย เงื่อนไขนี้เป็นไปตามวิธีการชดเชย ซึ่งขึ้นอยู่กับข้อเท็จจริงที่ว่าองค์ประกอบเชื่อมต่อแบบอนุกรมกับความต่างศักย์ภายนอก และเลือกอย่างหลังเพื่อไม่ให้มีกระแสไฟฟ้าในวงจร จากนั้นความต่างศักย์ภายนอกจะเท่ากับ EMF ของวงจร
เมื่อใช้วิธีการชดเชยคุณสามารถวัดค่าของ EMF ได้โดยตรง แต่นี่เป็นการดำเนินการที่ค่อนข้างซับซ้อนดังนั้นในทางปฏิบัติในห้องปฏิบัติการพวกเขาต้องการเปรียบเทียบ EMF ขององค์ประกอบที่กำลังศึกษากับ EMF ของมาตรฐานที่เรียกว่า (ปกติ) องค์ประกอบซึ่งมีการวัดอย่างระมัดระวังที่ T ที่แตกต่างกัน วิธีการเปรียบเทียบนี้ก็เป็นการชดเชยเช่นกัน
องค์ประกอบปกติพื้นฐานคือ องค์ประกอบเวสตันอิ่มตัว .
(วงจรวัด EMF - แยกกัน)
โครงสร้างของขอบเขตอิเล็กโทรด - สารละลาย. ชั้นไฟฟ้าสองชั้น .
เมื่อตัวนำชนิดที่ 1 สัมผัสกับอิเล็กโทรไลต์ a ไฟฟ้าสองชั้น . ตัวอย่างเช่น พิจารณาอิเล็กโทรดทองแดงที่แช่อยู่ในสารละลาย CuSO 4 ศักยภาพทางเคมีของไอออนทองแดงในโลหะที่ค่า T ที่กำหนดสามารถพิจารณาได้ว่าคงที่ ในขณะที่ศักยภาพทางเคมีของไอออนทองแดงในสารละลายขึ้นอยู่กับความเข้มข้นของเกลือ โดยทั่วไปศักยภาพทางเคมีเหล่านี้จะไม่เหมือนกัน
ให้ความเข้มข้นของ CuSO 4 เป็นเช่นนั้น > จากนั้น เมื่อโลหะถูกจุ่มลงในสารละลาย ไอออน Cu 2+ บางส่วนจากสารละลายจะถูกทำให้แห้งและถ่ายโอนไปยังโลหะ ทำให้เกิดประจุบวกขึ้น ประจุนี้จะป้องกันการถ่ายโอนไอออน Cu 2+ จากสารละลายไปยังโลหะ และจะทำให้เกิดชั้นของไอออน SO 4 2- แอนไอออนที่ดึงดูดใกล้กับอิเล็กโทรด ที่เรียกว่า สมดุลเคมีไฟฟ้า ซึ่งศักยภาพทางเคมีของไอออนในโลหะและในสารละลายจะแตกต่างกันตามขนาดของความต่างศักย์ของชั้นไฟฟ้าสองชั้น (DEL) ที่เกิดขึ้น:
ความต่างศักย์ไฟฟ้าและความต่างศักย์เคมีได้รับการชดเชยในสมดุลเคมีไฟฟ้า
ให้ความเข้มข้นของ CuSO 4 ต่ำขนาดนั้น< . В этом случае при погружении металла в раствор будет наблюдаться обратный процесс перехода ионов меди из кристаллической решетки металла в раствор и электрод окажется заряженным отрицательно. Этот заряд будет препятствовать дальнейшему переходу ионов Cu 2+ в раствор, установится новое электрохимическое равновесие.
คุณสามารถเลือกความเข้มข้นของอิเล็กโทรไลต์ที่มีศักยภาพทางเคมีของไอออนในโลหะและสารละลายเท่ากันได้ สารละลายของความเข้มข้นนี้เรียกว่า โซลูชั่นเป็นศูนย์ . เมื่อโลหะจุ่มอยู่ในสารละลายศูนย์ EDL จะเกิดขึ้นบนพื้นผิวของอิเล็กโทรด อย่างไรก็ตาม ในกรณีนี้ ความต่างศักย์ระหว่างโลหะกับสารละลายไม่เป็นศูนย์
จากข้อมูลของ Nernst แหล่งที่มาเดียวของ EMF ของเซลล์ไฟฟ้าเคมีคือ EMF บนพื้นผิวของอิเล็กโทรด Nernst กำหนดศักยภาพของโลหะในสารละลายศูนย์ว่าเป็นศักยภาพเป็นศูนย์สัมบูรณ์ ในผลงานของ A.N. Frumkin แสดงให้เห็นว่าแนวคิดของ Nernst นั้นไม่ถูกต้อง มีการทดลองพบว่า EMF ขององค์ประกอบที่ประกอบด้วยอิเล็กโทรดที่แตกต่างกันสองอิเล็กโทรดที่แช่อยู่ในสารละลายศูนย์จะแตกต่างจากศูนย์อย่างมาก (อาจมากกว่า 1 V) ศักยภาพของโลหะในสารละลายศูนย์เรียกว่า ศักยภาพการชาร์จเป็นศูนย์ ไม่สามารถพิจารณาได้ว่ามีศักยภาพเป็นศูนย์สัมบูรณ์
ทฤษฎีของ Helmholtz สองชั้นแบบควบแน่น. ทฤษฎีเชิงปริมาณแรกของโครงสร้างของ DEL ที่ส่วนต่อประสานของสารละลายโลหะถูกสร้างขึ้นโดย Helmholtz (1853) จากข้อมูลของ Helmholtz EDL สามารถเปรียบได้กับตัวเก็บประจุแบบแบน แผ่นหนึ่งซึ่งเกิดขึ้นพร้อมกับระนาบที่ผ่านประจุที่พื้นผิวในโลหะ และอีกแผ่นหนึ่งมีระนาบที่เชื่อมต่อศูนย์กลางของประจุของไอออนในสารละลาย ดึงดูดพื้นผิวโลหะด้วยแรงไฟฟ้าสถิต ความหนาสองชั้น ลเท่ากับรัศมีไอออน ร. ตามเงื่อนไขของความเป็นกลางทางไฟฟ้า จำนวนไอออนที่ดึงดูดกับพื้นผิวของโลหะจะต้องเป็นเช่นนั้น ประจุของพวกมันจะชดเชยประจุที่พื้นผิวของโลหะ เช่น
ทฤษฎีการควบแน่นสองชั้นทำให้สามารถรับค่าความจุ EDL ที่สอดคล้องกับการทดลองและความหนาที่เป็นไปได้ทางกายภาพของ EDL อย่างไรก็ตามไม่สามารถตีความความสม่ำเสมอของการทดลองได้หลายอย่าง: ค่าที่พบในการทดลองของศักย์ไฟฟ้า (x-potential) และการพึ่งพาความเข้มข้นของอิเล็กโทรไลต์, การเปลี่ยนแปลงเครื่องหมายของประจุบนพื้นผิวโลหะเมื่อมีสารลดแรงตึงผิว .
ทฤษฎี GUI สองชั้นแบบกระจาย- แชปแมน. ทฤษฎีของเฮล์มโฮลทซ์ไม่ได้คำนึงว่าคุณสมบัติของ DES เปลี่ยนแปลงไปตามความเข้มข้นของอิเล็กโทรไลต์ และ T. Gouy (1910) และ Chapman (1913) พยายามเชื่อมโยงความหนาแน่นประจุใน DES กับองค์ประกอบของสารละลาย พวกเขาคำนึงถึงว่านอกเหนือจากแรงไฟฟ้าสถิตที่เกิดขึ้นระหว่างโลหะกับไอออนแล้ว ไอออนยังได้รับผลกระทบจากแรงการเคลื่อนที่ของโมเลกุลความร้อนด้วย เมื่อใช้แรงทั้งสองนี้ ไอออนในสารละลายควรกระจายอย่างกระจายสัมพันธ์กับพื้นผิวโลหะ โดยความหนาแน่นประจุเชิงปริมาตรจะลดลงตามระยะห่างจากไอออน
Gouy และ Chapman เชื่อว่าไอออนถือเป็นจุดวัสดุที่ไม่มีปริมาตรเป็นของตัวเอง แต่มีประจุ และการกระจายตัวของไอออนในสนามประจุอิเล็กโทรดเป็นไปตามการกระจายตัวของ Boltzmann
ทฤษฎี Gouy-Chapman เห็นด้วยกับกฎของปรากฏการณ์ไฟฟ้าจลน์ได้ดีกว่าทฤษฎี Helmholtz หากเราถือว่าเริ่มจากระยะหนึ่ง ลไอออน 1 จะไม่เกาะติดกับพื้นผิวอิเล็กโทรดอย่างแน่นหนาอีกต่อไปด้วยการเคลื่อนที่สัมพัทธ์ของเฟสของแข็งและของเหลว ดังนั้นศักยภาพที่สอดคล้องกับระยะนี้จึงถือเป็นศักยภาพ x (x< j). Однако теория не объясняет изменение знака x-потенциала и перезарядку поверхности с изменением состава раствора. Кроме того, теория Гуи-Чапмана оказывается менее удовлетворительной, чем теория Гельмгольца, при использовании ее для количественных расчетов емкости ДЭС, т.к. она не учитывает собственного объема ионов, которые отождествляются с материальными точками.
ดังนั้น ทฤษฎี Gouy-Chapman จึงมีเหตุผลที่ดีที่สุด เมื่อทฤษฎี Helmholtz กลายเป็นว่าใช้ไม่ได้ และในทางกลับกัน ทฤษฎีหลังให้การบรรจบกันกับการทดลองได้ดีกว่า ในกรณีที่ทฤษฎีแรกให้ผลลัพธ์ที่ไม่ถูกต้อง ดังนั้น โครงสร้างของ DES จะต้องสอดคล้องกับแบบจำลองบางรุ่นที่เสนอโดย Helmholtz และ Gouy-Chapman สมมติฐานนี้จัดทำขึ้นโดยสเติร์น (1924) ในทฤษฎีการดูดซับของ DEL
ทฤษฎีการดูดซับที่เข้มงวด. สเติร์นเชื่อว่าไอออนบางส่วนยังคงอยู่ใกล้กับส่วนต่อประสานระหว่างโลหะกับอิเล็กโทรไลต์ ก่อตัวเป็นเฮล์มโฮลทซ์หรือแผ่นสองชั้นที่ควบแน่นซึ่งมีความหนาสอดคล้องกับรัศมีเฉลี่ยของไอออนอิเล็กโทรไลต์ ไอออนที่เหลือรวมอยู่ใน EDL จะถูกกระจายอย่างกระจัดกระจายโดยความหนาแน่นของประจุจะค่อยๆ ลดลง สำหรับส่วนที่แพร่กระจายของ EDL นั้น สเติร์น เช่นเดียวกับ Gouy ละเลยขนาดที่แท้จริงของไอออน นอกจากนี้ สเติร์นยังแนะนำว่าในส่วนที่หนาแน่นของ EDL ไอออนจะยังคงอยู่เนื่องจากไม่เพียงแต่แรงไฟฟ้าสถิตเท่านั้น แต่ยังรวมถึงแรงดูดซับจำเพาะด้วย เช่น แรงที่ไม่ใช่คูลอมบ์ ดังนั้นในสารละลายที่มีไอออนที่ออกฤทธิ์ที่พื้นผิว จำนวนของไอออนในส่วนที่มีความหนาแน่นของ EDL อาจเกินประจุของพื้นผิวโลหะได้ในระดับหนึ่ง ขึ้นอยู่กับคุณสมบัติของไอออนและประจุของโลหะ ดังนั้น ตามข้อมูลของสเติร์น ควรแยกแยะ DES สองรุ่น โดยรุ่นหนึ่งเกี่ยวข้องกับสารละลายของอิเล็กโทรไลต์ที่ไม่ใช้งานบนพื้นผิว และอีกรุ่นหนึ่งเกี่ยวข้องกับสารละลายที่มีไอออนดูดซับเป็นพิเศษ
ในทฤษฎีการดูดซับ ความเท่าเทียมกันยังคงอยู่:
Q M = q L = q 1 + q 2
ความหนาแน่นประจุที่ด้านสารละลาย q L ประกอบด้วยสองส่วน คือ ความหนาแน่นประจุในชั้น Helmholtz q 1 และความหนาแน่นประจุในชั้นกระจาย q 2
ทฤษฎีของสเติร์นช่วยให้เราสามารถกำหนดศักยภาพ x ว่าเป็นการลดลงที่อาจเกิดขึ้นในส่วนที่กระจัดกระจายของ EDL ซึ่งพันธะอันแน่นแฟ้นระหว่างโลหะกับไอออนได้สูญเสียไปแล้ว ด้วยคำจำกัดความนี้ ศักย์ x ไม่ควรตรงกับศักย์ Nerst ดังที่สังเกตได้จากการทดลอง ทฤษฎีของสเติร์นสามารถอธิบายการอัดประจุใหม่ของพื้นผิววัตถุแข็งได้
ที่ความเข้มข้นเพียงเล็กน้อย ประจุทั้งหมดในสารละลายจะถูกกระจายอย่างกระจัดกระจาย และโครงสร้างของ EDL อธิบายไว้ในทฤษฎี Gouy-Chapman ในทางตรงกันข้าม ในการแก้ปัญหาแบบเข้มข้น โครงสร้างของ DES จะเข้าใกล้แบบจำลองที่ Helmholtz เสนอ ในบริเวณความเข้มข้นเฉลี่ย โดยที่ x มีขนาดเทียบเคียงได้กับ RT/F การขึ้นอยู่กับความเข้มข้นสามารถแสดงได้ด้วยสมการโดยประมาณ:
สำหรับค่าบวก x: x = B - lnกับ
สำหรับค่าลบของ x: x = B¢ + lnกับ
ทฤษฎีของสเติร์นให้ภาพ DEL ที่ถูกต้องในเชิงคุณภาพ การกำหนดความจุไฟฟ้าโดยใช้แบบจำลอง Stern นั้นสอดคล้องกับประสบการณ์ทั้งในแง่ของค่าความจุและในลักษณะของการขึ้นอยู่กับศักย์ไฟฟ้าของอิเล็กโทรดและความเข้มข้นของสารละลาย แต่ทฤษฎีของสเติร์นไม่ได้ปราศจากข้อบกพร่อง ซึ่งรวมถึงความเป็นไปไม่ได้ที่จะอธิบายเชิงปริมาณของเส้นโค้งความจุ โดยเฉพาะอย่างยิ่งเมื่อเคลื่อนออกจากศักย์ไฟฟ้าเป็นศูนย์
การพัฒนาเพิ่มเติมของทฤษฎี DES STANDING. มีความพยายามหลายครั้งในการพัฒนาทฤษฎี DES ที่มีความสอดคล้องกับข้อมูลการทดลองในเชิงปริมาณ (Rice, Frumkin และคณะ, Bockris, Devanathan, Esin, Muller, Parsons, Ershler เป็นต้น) แบบจำลองที่ได้รับการยอมรับอย่างกว้างขวางที่สุดคือ Graham (1947) ตามข้อมูลของ Graham การเคลือบ DES ในสารละลายไม่ได้ประกอบด้วยสองส่วน แต่มีสามส่วน ประการแรกนับจากพื้นผิวโลหะเรียกว่าระนาบเฮล์มโฮลทซ์ภายใน ประกอบด้วยไอออนที่ออกฤทธิ์ที่พื้นผิวเท่านั้น (ประจุของระนาบคือ q 1) หรือหากไม่อยู่ในสารละลาย โมเลกุลของตัวทำละลาย (q 1 = 0) ศักยภาพที่เกี่ยวข้องกับการแก้ปัญหาแสดงด้วย y 1 อันถัดไปที่นำออกจากพื้นผิวโลหะในระยะห่างที่ไอออน (ศูนย์ประจุของพวกมัน) สามารถเข้าใกล้ได้ เรียกว่าระนาบเฮล์มโฮลทซ์ส่วนนอก ประจุรวมของมันคือ q 2 และศักยภาพของเครื่องบินคือ y 2 ด้านหลังระนาบเฮล์มโฮลทซ์ด้านนอกมีชั้นกระจายที่มีศักยภาพเปลี่ยนแปลงตั้งแต่ y 2 ถึงศูนย์ และมีความหนาแน่นประจุตรงกับ q 2
แบบจำลองของ Graham สะท้อนถึงคุณสมบัติหลักและคุณลักษณะของโครงสร้าง DES โลหะอิเล็กโทรไลต์ ช่วยให้คุณสามารถคำนวณกราฟค่าความจุไฟฟ้าแบบดิฟเฟอเรนเชียลสำหรับความเข้มข้นใดๆ ของอิเล็กโทรไลต์ที่ระบุได้ หากมีกราฟทดลองสำหรับสารละลายอย่างน้อยหนึ่งค่า อย่างไรก็ตาม โมเดลนี้ไม่ครอบคลุมทุกแง่มุมของปัญหา
เมื่อกระแสไฟฟ้าไหลผ่านสารละลาย กระแสจะไหลบนพื้นผิวของอิเล็กโทรด ปฏิกิริยาเคมีไฟฟ้าซึ่งมาพร้อมกับการไหลของอิเล็กตรอนเข้าหรือออกจากอิเล็กโทรด ในกระบวนการย้อนกลับ ปฏิกิริยาเคมีไฟฟ้าที่เกิดขึ้นที่ส่วนต่อประสานระหว่างตัวนำชนิดที่หนึ่งและชนิดที่สองนำไปสู่การสร้างกระแสไฟฟ้า
กระบวนการเคมีไฟฟ้าแตกต่างจากปฏิกิริยาเคมีทั่วไปในคุณสมบัติหลายประการ
ปฏิกิริยาเคมีจะเกิดขึ้นได้ก็ต่อเมื่ออนุภาคที่ทำปฏิกิริยาชนกัน เมื่อสัมผัสกัน อิเล็กตรอนจะถ่ายโอนจากอนุภาคหนึ่งไปยังอีกอนุภาคหนึ่งได้ การเปลี่ยนแปลงดังกล่าวจะเกิดขึ้นจริงหรือไม่นั้นขึ้นอยู่กับพลังงานของอนุภาคและการวางแนวซึ่งกันและกัน พลังงานกระตุ้นขึ้นอยู่กับลักษณะของปฏิกิริยาเคมี และสำหรับปฏิกิริยาไอออนิกมักจะมีค่าต่ำ เส้นทางการเปลี่ยนผ่านของอิเล็กตรอนนั้นสั้นมาก ซึ่งเป็นคุณลักษณะของปฏิกิริยาเคมีด้วย การชนกันของอนุภาคสามารถเกิดขึ้นได้ที่จุดใดๆ ของพื้นที่ปฏิกิริยาที่ตำแหน่งร่วมกันที่แตกต่างกัน ดังนั้น การเปลี่ยนผ่านทางอิเล็กทรอนิกส์จึงสามารถเกิดขึ้นในทิศทางใดก็ได้ เช่น คุณสมบัติของกระบวนการทางเคมีคือการสุ่มของการชนและการขาดทิศทางของการเปลี่ยนผ่านทางอิเล็กทรอนิกส์ เป็นผลให้ผลกระทบที่มีพลังของปฏิกิริยาเคมีปรากฏอยู่ในรูปของความร้อนเป็นหลัก (สามารถขยายตัวเล็กน้อยได้เช่นกัน)
เพื่อให้การเปลี่ยนแปลงพลังงานที่สอดคล้องกับการเปลี่ยนแปลงทางเคมีปรากฏอยู่ในรูปของพลังงานไฟฟ้านั่นคือ เพื่อให้กระบวนการไฟฟ้าเคมีดำเนินต่อไปได้ จำเป็นต้องเปลี่ยนสภาวะของปฏิกิริยา
พลังงานไฟฟ้าสัมพันธ์กับเส้นทางของกระแสไฟฟ้าเสมอเช่น การไหลของอิเล็กตรอนไปในทิศทางใดทิศทางหนึ่ง ดังนั้นปฏิกิริยาจะต้องดำเนินการในลักษณะที่การเปลี่ยนทางอิเล็กทรอนิกส์ไม่สุ่ม แต่เกิดขึ้นในทิศทางเดียวและเส้นทางของมันจะต้องมีขนาดใหญ่กว่าขนาดอะตอมอย่างมาก ดังนั้นในกระบวนการเคมีไฟฟ้า การเปลี่ยนผ่านของอิเล็กตรอนจากผู้เข้าร่วมรายหนึ่งไปยังอีกรายหนึ่งจะต้องเกิดขึ้นในระยะห่างพอสมควร ซึ่งจำเป็นต้องมีการแยกเชิงพื้นที่ของผู้เข้าร่วมปฏิกิริยา อย่างไรก็ตาม การแยกพื้นที่เพียงอย่างเดียวไม่เพียงพอ เนื่องจากจะทำให้ปฏิกิริยาหยุดลง
ในการดำเนินการกระบวนการเคมีไฟฟ้าจำเป็นต้องมีเงื่อนไขเพิ่มเติม: อิเล็กตรอนจะต้องถูกแยกออกจากอนุภาคบางส่วนและถ่ายโอนไปยังอนุภาคอื่นด้วยวิธีทั่วไปแบบเดียวกัน สิ่งนี้สามารถทำได้โดยแทนที่การสัมผัสโดยตรงระหว่างผู้เข้าร่วมในการทำปฏิกิริยาด้วยการสัมผัสกับโลหะสองชนิดที่เชื่อมต่อถึงกันด้วยตัวนำโลหะ เพื่อให้การไหลของอิเล็กตรอนเป็นไปอย่างต่อเนื่องจำเป็นต้องให้แน่ใจว่ากระแสไฟฟ้าผ่านพื้นที่ปฏิกิริยาซึ่งโดยปกติผู้เข้าร่วมในปฏิกิริยาเคมีไฟฟ้าจะดำเนินการด้วยตนเอง (หากอยู่ในสถานะแตกตัวเป็นไอออน) หรือ ด้วยสารประกอบพิเศษที่มีค่าการนำไฟฟ้าไอออนิกสูง
อุปกรณ์สำหรับผลิตพลังงานไฟฟ้าผ่านปฏิกิริยาเคมีไฟฟ้าเรียกว่า เคมีไฟฟ้า(หรือ กัลวานิก)องค์ประกอบ. องค์ประกอบไฟฟ้าเคมีที่ง่ายที่สุดประกอบด้วยอิเล็กโทรดโลหะสองตัว (ตัวนำชนิดที่หนึ่ง) แช่อยู่ในสารละลายอิเล็กโทรไลต์ (ตัวนำชนิดที่สอง)
หากเมื่อกระแสไฟฟ้าไหลไปในทิศทางที่แตกต่างกันปฏิกิริยาเดียวกันเกิดขึ้นบนพื้นผิวของอิเล็กโทรด แต่ไปในทิศทางตรงกันข้ามอิเล็กโทรดดังกล่าวรวมถึงองค์ประกอบไฟฟ้าเคมีที่ประกอบด้วยพวกมันจะถูกเรียกว่า ย้อนกลับได้. ตัวอย่างขององค์ประกอบที่พลิกกลับได้คือองค์ประกอบ Daniel–Jacobi
(–) สังกะสี | ZnSO 4 โซลูชัน || CuSO 4 สารละลาย | ลูกบาศ์ก(+)
เมื่อองค์ประกอบดังกล่าวทำงาน ปฏิกิริยาเคมีไฟฟ้าจะเกิดขึ้นที่อิเล็กโทรด:
สังกะสี สังกะสี 2 + + 2e
Cu 2 + + 2eCu
สมการปฏิกิริยาโดยรวมในองค์ประกอบสามารถแสดงได้ดังนี้
สังกะสี + Cu 2 + Zn 2 + + Cu
เมื่อกระแสไฟฟ้าจำนวนเล็กน้อยจากแหล่งภายนอกถูกส่งผ่านองค์ประกอบ ปฏิกิริยาเหล่านี้จะดำเนินไปในทิศทางตรงกันข้าม
ตัวอย่าง กลับไม่ได้องค์ประกอบคือองค์ประกอบโวลตา
(–) สังกะสี | H2SO4 | ลูกบาศ์ก(+)
เมื่อองค์ประกอบดังกล่าวทำงาน ปฏิกิริยาต่อไปนี้จะเกิดขึ้นที่อิเล็กโทรด:
สังกะสี สังกะสี 2 + + 2e
2H + + 2อีH 2 ,
และปฏิกิริยาในองค์ประกอบนั้นแสดงด้วยสมการ
สังกะสี + 2H + สังกะสี 2+ + H 2
เมื่อกระแสไฟฟ้าถูกส่งผ่านจากแหล่งภายนอก ปฏิกิริยาอื่นๆ จะเกิดขึ้นที่อิเล็กโทรด:
ลูกบาศ์ก ลูกบาศ์ก 2 + + 2e,
เหล่านั้น. ในเซลล์ไฟฟ้าเคมี ทองแดงจะละลายในกรดซัลฟิวริกโดยปล่อยไฮโดรเจน:
Cu + 2H + Cu 2 + + H 2
ลักษณะที่สำคัญที่สุดของเซลล์ไฟฟ้าเคมีก็คือ แรงเคลื่อนไฟฟ้า(อีเอ็มเอฟ) อี– ความต่างศักย์ขององค์ประกอบที่เปิดอย่างเหมาะสม เช่น ความต่างศักย์ระหว่างปลายตัวนำของวัสดุชนิดเดียวกันชนิดแรกที่เชื่อมต่อกับอิเล็กโทรดของเซลล์กัลวานิก กล่าวอีกนัยหนึ่ง EMF คือความต่างศักย์ภายใต้สภาวะสมดุลเมื่อไม่มีกระแสไฟฟ้าไหลในวงจร หากคุณปิดอิเล็กโทรด กระแสไฟฟ้าจะไหลในวงจร และความต่างศักย์แสดงถึง แรงดันไฟฟ้าองค์ประกอบไฟฟ้าเคมีที่แตกต่างจาก EMF ตามปริมาณแรงดันไฟฟ้าตกคร่อมความต้านทานภายในขององค์ประกอบ
จุดประสงค์ขององค์ประกอบไฟฟ้าเคมีคือเพื่อส่งเสริมการเคลื่อนที่ของอิเล็กตรอนในวงจรภายนอกซึ่งมีองค์ประกอบที่มีประโยชน์รวมอยู่ด้วย
โหลด ดังนั้น เพื่อดำเนินงาน เซลล์เชื้อเพลิงจะต้องมีแหล่งกำเนิดและแหล่งเก็บอิเล็กตรอน (รูปที่ 7.1)
ปฏิกิริยาที่เกิดขึ้นในเซลล์ไฟฟ้าเคมีเรียกว่าปฏิกิริยารีดักชัน เนื่องจากคำว่า "ออกซิเดชัน" สอดคล้องกับกระบวนการปล่อยอิเล็กตรอน และคำว่า "การรีดิวซ์" สอดคล้องกับกระบวนการปล่อยอิเล็กตรอน
ความหมายของแนวคิดทางวิทยาศาสตร์ที่ล้าสมัยจำนวนมากมักไม่ชัดเจนจากชื่อ คำว่า "ออกซิเดชัน" และ "การลดลง" จำเป็นต้องมีคำอธิบาย ในภาษาตะวันออกเฉียงใต้ ออกซิเจน (ออกซิเจน) มาจากรากภาษาละติน oxus (เปรี้ยวหรือร้อน, คม) และหมายถึง "ผู้ผลิตกรด" ชื่อนี้ถูกใช้ครั้งแรกในงานของ Morveau และ Lavoisier “Nomenclature Chimique” ซึ่งตีพิมพ์ในปี พ.ศ. 2330 ในเวลานั้นนักเคมียึดติดกับความเชื่อที่ผิด ออกซิเจนเป็นองค์ประกอบหลักที่อยู่ในกรด เมื่อกรดละลายในน้ำ อะตอมไฮโดรเจนส่วนหนึ่งที่อยู่ในองค์ประกอบจะสูญเสียอิเล็กตรอนไป น้ำจะกลายเป็นกรด และไฮโดรเจนจะถูกออกซิไดซ์ ในทางวิเคราะห์ ปฏิกิริยาใดๆ ที่เกี่ยวข้องกับกระบวนการสูญเสียอิเล็กตรอนเรียกว่าออกซิเดชัน ปฏิกิริยาย้อนกลับ - ปฏิกิริยาการจับอิเล็กตรอน - เรียกว่า ■กลายเป็น
ไหล
เจอิเล็กตรอน
แหล่งกำเนิดอิเล็กตรอน >
(ออกซิเดชัน)
เชิงบวก< -
ทิศทางของกระแสไฟฟ้า รูปที่. 7.1. เซลล์ไฟฟ้าเคมีจะต้องมีแหล่งกำเนิดและการจมของอิเล็กตรอน
ในเซลล์ไฟฟ้าเคมี ปฏิกิริยาโดยรวมจะถูกแบ่งออกเป็นปฏิกิริยาขั้นกลางสองปฏิกิริยาที่เกิดขึ้นในพื้นที่ที่แยกจากกันของอุปกรณ์ พื้นที่เหล่านี้ถูกหลอมรวมโดยอิเล็กโทรไลต์ซึ่งเป็นตัวนำไอออน แต่นำอิเล็กตรอนที่ปล่อยออกมาในปฏิกิริยาออกซิเดชันระดับกลาง อิเล็กตรอนสามารถเข้าสู่บริเวณที่เกิดปฏิกิริยารีดักชัน แต่ผ่านวงจรภายนอก ดังนั้นกระแสไฟฟ้าจึงเกิดขึ้นในวงจรภายนอกและเซลล์ไฟฟ้าเคมีเป็นแหล่งกำเนิด - นี่คือจุดประสงค์ของมัน ทิศทางบวกของไฟฟ้า a ในวงจรภายนอกถูกตกลงให้เป็นทิศทางจากบริเวณรีดิวซ์ออกซิเดชันขององค์ประกอบ - กระแสไฟฟ้าออกจากองค์ประกอบของบริเวณรีดักชัน ซึ่งก็คือแคโทดของเชื้อเพลิง - o องค์ประกอบ และ เข้าสู่บริเวณออกซิเดชันซึ่งก็คือขั้วบวก เช่นเดียวกับแหล่งไฟฟ้าอื่นๆ แคโทดมีประจุบวก
อิเล็กโทรดที่ใช้งานอยู่และแอโนดเป็นอิเล็กโทรดที่มีประจุลบ เมื่อพูดถึงผู้ใช้พลังงานไฟฟ้า (โหลด) ชื่อจะเปลี่ยนไปตรงกันข้าม ในบทนำของช. 6 จะมีการกล่าวถึงรายละเอียดเพิ่มเติมเกี่ยวกับคำว่า "แอโนด" และ "แคโทด"
เป็นตัวอย่างของเซลล์ไฟฟ้าเคมี ให้พิจารณาเมมเบรนที่ทำหน้าที่เป็นอิเล็กโทรไลต์ ปล่อยให้พื้นผิวด้านข้างด้านใดด้านหนึ่งของเมมเบรนสัมผัสกับไฮโดรเจน ภายใต้สภาวะปกติ ก๊าซจะประกอบด้วยโมเลกุลไฮโดรเจนเป็นส่วนใหญ่ แต่โมเลกุลจำนวนเล็กน้อยอาจแยกตัวออกเป็นอะตอม
และอะตอมบางส่วนจะออกซิไดซ์ (แตกตัวเป็นไอออน) กล่าวคือ สูญเสียอิเล็กตรอนไป
ยังไม่มีข้อความ -> Nt + อี~
เนื่องจากเมมเบรนไม่สามารถซึมผ่านของอิเล็กตรอนได้ พวกมันทั้งหมดจะยังคงอยู่ที่ด้านใดด้านหนึ่งของเมมเบรน ในขณะที่ไอออนที่ก่อตัวจะผ่านส่วนต่าง ผ่านเมมเบรนพวกเขาจะพบว่าตัวเองอยู่อีกด้านหนึ่ง ในกรณีนี้ ไอออนมีประจุบวก ดังนั้นพื้นผิวของเมมเบรนซึ่งมีไฮโดรเจนอยู่จะมีประจุลบเนื่องจากมีอิเล็กตรอนส่วนเกินสะสมอยู่ และพื้นผิวด้านตรงข้ามจะมีประจุบวกเนื่องจากประจุบวก ไอออนที่ปรากฏเนื่องจากการแพร่กระจาย สนามไฟฟ้าทำให้ไอออนบางส่วนเคลื่อนที่ในทิศทางตรงกันข้ามกับพื้นผิว "ไฮโดรเจน" ของเมมเบรน สมดุลไดนามิกในระบบที่พิจารณาจะถูกสร้างขึ้นเมื่อการไหลของไอออนแพร่กระจายอยู่ที่ st ฉันเท่ากับกระแสย้อนกลับ
ตอนนี้ เรามาทาผงนำไฟฟ้ากับพื้นผิวทั้งสองของเมมเบรน ซึ่งจะส่งผลให้มีชั้นสื่อไฟฟ้าที่มีรูพรุน 2 ชั้นซึ่งจะทำหน้าที่เป็นอิเล็กโทรด มาเชื่อมต่อเครื่องทำความร้อนภายนอก1 เข้ากับอิเล็กโทรด เพื่อให้แน่ใจว่ามีการเชื่อมต่อทางไฟฟ้าของอิเล็กโทรด โหลดความต้านทานต่อ Rl ที่สำคัญ ไอออนไม่สามารถเคลื่อนที่ในวงจรภายนอกได้ และอิเล็กตรอนเริ่มเคลื่อนที่จากส่วน "ไฮโดรเจน" ซึ่งมีส่วนเกินไปยังด้านตรงข้ามของเมมเบรน ทำให้เกิดกระแสไฟฟ้าในวงจรภายนอก ดังในรูป 7.2. ปฏิกิริยาที่เราสนใจเกิดขึ้นที่อิเล็กโทรด "น้ำ" และอธิบายไว้ในสมการ
2H2 -> 4H+ + 4e (ปฏิกิริยาขั้วบวก)
รูปแบบการทำงานขององค์ประกอบที่อธิบายไว้มีข้อเสียเปรียบที่สำคัญประการหนึ่ง: มันขัดแย้งกับกฎข้อแรกของอุณหพลศาสตร์ แท้จริงแล้วเมื่อกระแสไฟฟ้าไหลผ่านโหลดภายนอก ปริมาณของกระแสไฟฟ้าจะถูกปล่อยออกมา ซึ่งปริมาณดังกล่าวจะถูกกำหนดโดยผลิตภัณฑ์ I2RL ขณะเดียวกันก็มีการไฟฟ้า
เข้าสู่บริเวณแคโทดรวมกับไอออนของ H+ ซึ่งกระจายผ่านเยื่อหุ้มเซลล์ เกิดเป็นอะตอมไฮโดรเจน ทำให้เกิดโมเลกุลของก๊าซ H2 ที่ใช้เป็น “เชื้อเพลิง” ในที่สุด หากดำเนินการตามโครงการที่กำหนด เราก็จะได้รับความร้อนโดยไม่สิ้นเปลืองเชื้อเพลิงแต่อย่างใด
วงจรภายนอกเป็นเพียงเส้นทางการเคลื่อนที่ของอิเล็กตรอน แต่ในตัวมันเองไม่สามารถเป็นสาเหตุของการเกิดกระแสไฟฟ้าได้ เช่นเดียวกับถ้าคุณลดปลายท่อด้านหนึ่งลงในทะเลสาบ น้ำจะไม่ไหลเข้าไปในท่อ เพื่อให้น้ำไหลได้ ปลายอีกด้านจะต้องอยู่ใต้ผิวน้ำ ในทำนองเดียวกัน เพื่อที่จะแนะนำกระแสไฟฟ้าในวงจรภายนอก จำเป็นต้องลดศักย์โรโมไดนามิกส์ในบริเวณแคโทด วิธีที่ง่ายที่สุดในการทำเช่นนี้คือการเติมออกซิเจน ซึ่งเป็นโมเลกุลที่รวมกับอิเล็กตรอนและไอออน ส่งผลให้เกิดการก่อตัวของน้ำ:
4е“ + 4Н+ + 02 -» 2Н20 (ปฏิกิริยาแคโทด) (4)
ปฏิกิริยานี้เป็นปฏิกิริยาคายความร้อน กล่าวคือ เกิดขึ้นจากการปล่อยพลังงานไฟฟ้าเป็นส่วนใหญ่ ไม่ใช่พลังงานความร้อน ซึ่งเกิดขึ้นเมื่อเผาไหม้ไฮโดรเจน แน่นอนว่าพลังงานนี้เองที่ให้พลังงานแก่องค์ประกอบเชื้อเพลิง
แผนภาพอธิบายของเซลล์ไฟฟ้าเคมีแสดงไว้ในรูปที่ 1 7.2.
ภายใต้สภาวะปกติ สัดส่วนของโมเลกุลไฮโดรเจนที่กระจาย* ทั่วทั้งบริเวณจะมีน้อย สามารถเพิ่มขึ้นได้เล็กน้อยโดยการเปลี่ยนพารามิเตอร์ทางกายภาพ
เมตรตามหลักการของเลอ ชาเตอลิเยร์ เช่น โดยการเพิ่มอุณหภูมิของระบบ ระดับการแยกตัวของโมเลกุลไฮโดรเจนสามารถเพิ่มขึ้นได้1 โดยใช้การกระทำของตัวเร่งปฏิกิริยา
ปฏิกิริยาสมบูรณ์ที่เกิดขึ้นในเซลล์ไฟฟ้าเคมีอธิบายได้ด้วยสมการ:
2H2 + 02 -> 2H?0
เซลล์ไฟฟ้าเคมีประดิษฐ์โดย Alessandro Volta (1745-1 ในปี 1800) เป็นอุปกรณ์แรกที่ผลิตกระแสไฟฟ้าในโหมดต่อเนื่อง ประกอบด้วยฐานสังกะสีและเงิน (หรือทองแดง) แยกจากกันด้วยแผ่นกระดาษแช่ในสารละลายเกลือ อิเล็กโทรดขององค์ประกอบหนึ่งจะเชื่อมต่อโดยตรงกับอิเล็กโทรด ZINC ของอีกองค์ประกอบหนึ่งโดยการเชื่อมต่อองค์ประกอบต่างๆ แบบอนุกรม
เซลล์ไฟฟ้าเคมีโวลตาสามารถสร้างขึ้นได้โดยการจุ่มอิเล็กโทรดแบบเชื่อมและอิเล็กโทรดทองแดงในสารละลายเจือจาง (เช่น 10%) ของกรดนี้ สังกะสีจะออกซิไดซ์:
สังกะสี -> Zr,++ + 2e >
เป็นผลให้อิเล็กตรอนอิสระเกิดขึ้น ละลายไอออนสังกะสีในน้ำ กรดซัลฟิวริกเป็นกรดแก่1) แยกตัวออกเป็นไอออน:
H2S04 - "2Н+ + SOf"
ซิงค์ไอออนรวมกับซัลเฟตไอออนเพื่อสร้างซิงค์ซัลเฟต พอนที่อยู่ในรูปของไฮโดรเนียมไอออน H" (H20)x จะเคลื่อนที่ผ่านอิเล็กโทรไลต์ไปยังอิเล็กโทรด ซึ่งจะถูกปล่อยออกมาในรูปของฟองก๊าซ (เมื่อไฮโดรเนียมไอออนจับอิเล็กตรอนที่มาจากภายนอกอิเล็กโทรดไปยังอิเล็กโทรด II วงจรไฟฟ้า) ในทางปฏิบัติเซลล์ไฟฟ้าเคมีชนิดนี้แทบไม่มีประโยชน์เนื่องจากอิเล็กโทรดทองแดงจะถูกปกคลุมไปด้วยฟองไฮโดรเจนที่ติดอยู่กับพื้นผิวอย่างรวดเร็วอย่างรวดเร็วซึ่งจะช่วยลดการไหลของไอออนที่เข้าสู่อิเล็กโทรดได้อย่างมาก สิ่งที่เรียกว่า "เซลล์แห้ง" ใช้วิธีการบางอย่างเพื่อป้องกันการก่อตัวของชั้นก๊าซที่เป็นฉนวนบนพื้นผิว รีเอเจนต์เคมีที่ใช้ในการดูดซับไฮโดรเจนเรียกว่าดีโพลาไรเซอร์ โดยปกติแล้วรีเอเจนต์ตัวหนึ่งจะใช้
"> ความแรงของกรดถูกกำหนดโดยระดับการแยกตัวของโมเลกุลในสารละลายที่เป็นน้ำ กรด C แยกตัวออกเป็นไอออน H+ และ C1_ อย่างสมบูรณ์ - นี่คือกรดแก่ กรดซัลฟูริกค่อนข้างอ่อนกว่า แต่ก็เป็นของกรดแก่ด้วย . น่าแปลกที่กรดไฮโดรฟลูออริกแม้จะมีฤทธิ์กัดกร่อนสูง แต่ก็เป็นกรดอ่อน ๆ ไม่ใช่สารละลายที่อุณหภูมิห้อง สัดส่วนของ H+ ไอออนจากความเข้มข้นรวมของโมเลกุลนิวตร้า HF นั้นน้อยกว่า 3%
น้ำดีอาจเกิดปฏิกิริยาออกซิเดชั่นได้ง่าย สังกะสีมักใช้บ่อยที่สุด โปรดทราบว่าอิเล็กโทรดทองแดงในองค์ประกอบ Volta จะไม่เกิดปฏิกิริยาเคมี ดังนั้นจึงไม่ได้ใช้ทองแดง
จนกระทั่งเมื่อไม่นานมานี้ แบตเตอรี่กัลวานิกราคาไม่แพงใช้เซลล์ Leclanchet ซึ่งขั้วบวกทำจากสังกะสีและแคโทดทำจากแท่งกราไฟท์ที่เคลือบด้วยชั้นแมงกานีสไดออกไซด์ที่เป็นผงด้วยการเติมกราไฟท์ (เพื่อเพิ่มการนำไฟฟ้า) แมงกานีสไดออกไซด์ดูดซับไฮโดรเจนที่ปล่อยออกมา ซึ่งป้องกันการก่อตัวของชั้นก๊าซบนพื้นผิวแคโทด แอมโมเนียมคลอไรด์ถูกใช้เป็นอิเล็กโทรไลต์ แบตเตอรี่สมัยใหม่ (อัลคาไลน์) ใช้อิเล็กโทรไลต์อัลคาไลน์
หากสังกะสีที่ใช้สำหรับวัสดุแอโนดมีความบริสุทธิ์อย่างแน่นอน สังกะสีจะถูกใช้เฉพาะเมื่อกระแสไฟฟ้าไหลผ่านองค์ประกอบเท่านั้น การปรากฏตัวของสิ่งเจือปนทำให้เกิดการกัดกร่อนของอิเล็กโทรดแม้ว่าจะไม่ได้ใช้งานองค์ประกอบก็ตาม (สิ่งเจือปนจะก่อตัวเป็นเซลล์ไฟฟ้าเคมีขนาดเล็กจำนวนมากภายในวัสดุอิเล็กโทรด) เพื่อให้แน่ใจว่าแบตเตอรี่ดังกล่าวมีอายุการเก็บรักษายาวนาน แอโนดจึงทำจากโลหะผสมของสังกะสีและปรอท (ผสมกัน) ปัจจุบันเซลล์ Leclanche ถูกแทนที่ด้วยแบตเตอรี่อัลคาไลน์เกือบทั้งหมดแล้ว
ปฏิกิริยาเคมีหลายอย่างเกิดขึ้นเฉพาะเมื่อมีการจ่ายพลังงานจากภายนอกเท่านั้น มักดำเนินการในเซลล์อิเล็กโทรไลต์ (อิเล็กโทรไลเซอร์) บนอิเล็กโทรดที่เชื่อมต่อกับแหล่งจ่ายกระแสภายนอก การศึกษาปฏิกิริยาเหล่านี้ให้ข้อมูลเกี่ยวกับธรรมชาติและคุณสมบัติของสารต่างๆ และยังช่วยให้ได้สารประกอบเคมีใหม่โดยใช้การสังเคราะห์ด้วยไฟฟ้า กระบวนการไฟฟ้าเคมีมีการใช้กันอย่างแพร่หลายในอุตสาหกรรม ตัวอย่าง ได้แก่ การผลิตคลอรีนและอะลูมิเนียม การชุบด้วยไฟฟ้า และการสกัดด้วยไฟฟ้า เซลล์กัลวานิกซึ่งแปลงพลังงานเคมีเป็นพลังงานไฟฟ้า เป็นพื้นฐานของแหล่งที่มาของกระแส - แบตเตอรี่และตัวสะสมพลังงาน เช่นเดียวกับเซลล์เชื้อเพลิง เคมีไฟฟ้ายังศึกษาปรากฏการณ์ทางไฟฟ้าอื่นๆ ด้วย เช่น พฤติกรรมของไอออนในสารละลายอิเล็กโทรไลต์และการผ่านของกระแสผ่านสารละลายดังกล่าว การแยกไอออนในสนามไฟฟ้า (อิเล็กโทรโฟเรซิส) การกัดกร่อนและการทู่ของโลหะ ผลกระทบทางไฟฟ้าในระบบชีวภาพ (ชีวไฟฟ้าเคมี) กระบวนการโฟโตอิเล็กโตรเคมี (ผลของแสงต่อปฏิกิริยาเคมีไฟฟ้าในเซลล์)
การอ้างอิงทางประวัติศาสตร์
เป็นไปได้ที่จะดำเนินการวิจัยไฟฟ้าเคมีอย่างเป็นระบบหลังจากสร้างแหล่งกระแสไฟฟ้าที่คงที่และมีพลังเพียงพอเท่านั้น แหล่งที่มาดังกล่าวปรากฏขึ้นในช่วงเปลี่ยนศตวรรษที่ 18–19 อันเป็นผลมาจากผลงานของ L. Galvani และ A. Volta ในขณะที่ศึกษาการทำงานทางสรีรวิทยาของกบ กัลวานีได้สร้างวงจรเคมีไฟฟ้าโดยไม่ได้ตั้งใจซึ่งประกอบด้วยโลหะสองชนิดที่แตกต่างกันและกล้ามเนื้อของขากบที่ผ่าออก เมื่ออุ้งเท้าซึ่งยึดไว้ด้วยที่ยึดทองแดงถูกสัมผัสด้วยลวดเหล็กที่เชื่อมต่อกับที่ยึดเช่นกัน กล้ามเนื้อก็หดตัว การหดตัวที่คล้ายกันเกิดขึ้นภายใต้อิทธิพลของการปล่อยกระแสไฟฟ้า กัลวานีอธิบายปรากฏการณ์นี้โดยการมีอยู่ของ "ไฟฟ้าจากสัตว์" โวลตาตีความการทดลองเหล่านี้ให้แตกต่างออกไป ซึ่งเชื่อว่ากระแสไฟฟ้าเกิดขึ้น ณ จุดที่โลหะทั้งสองสัมผัสกัน และการหดตัวของกล้ามเนื้อกบเป็นผลมาจากการที่กระแสไฟฟ้าผ่านเข้าไป กระแสไฟฟ้ายังเกิดขึ้นเมื่อวางวัสดุที่เป็นรูพรุน (ผ้าหรือกระดาษ) แช่ในน้ำเกลือไว้ระหว่างแผ่นโลหะสองแผ่น เช่น สังกะสีและทองแดง และวงจรปิด ด้วยการเชื่อมต่อ "องค์ประกอบ" 15-20 เหล่านี้เข้าด้วยกันแบบอนุกรม โวลตาในปี 1800 ได้สร้างแหล่งกำเนิดกระแสเคมีแห่งแรก - "คอลัมน์โวลตาอิก"
อิทธิพลของไฟฟ้าที่มีต่อระบบเคมีเป็นที่สนใจของนักวิทยาศาสตร์หลายคนในทันที ในปี 1800 W. Nicholson และ A. Carlyle รายงานว่าน้ำสลายตัวเป็นไฮโดรเจนและออกซิเจนเมื่อมีกระแสไฟฟ้าไหลผ่านโดยใช้สายแพลตตินัมและทองคำที่เชื่อมต่อกับ "เสาโวลตาอิก" การศึกษาเคมีไฟฟ้ายุคแรกที่สำคัญที่สุดคืองานของนักเคมีชาวอังกฤษ H. Davy ในปี ค.ศ. 1807 เขาได้แยกธาตุโพแทสเซียมโดยการส่งกระแสผ่านโพแทสเซียมไฮดรอกไซด์ที่เป็นของแข็งที่ชุบน้ำเล็กน้อย แหล่งกำเนิดแรงดันไฟฟ้าคือแบตเตอรี่เซลล์กัลวานิก 100 เซลล์ ได้รับโซเดียมโลหะในลักษณะเดียวกัน ต่อมาเดวีได้แยกแมกนีเซียม แคลเซียม สตรอนเซียม และแบเรียมโดยอิเล็กโทรไลซิสโดยใช้อิเล็กโทรดปรอท
เอ็ม. ฟาราเดย์ ผู้ช่วยของเดวี ตรวจสอบความสัมพันธ์ระหว่างปริมาณไฟฟ้า (เวลาปัจจุบัน เวลา) ที่ไหลผ่านส่วนต่อประสานของอิเล็กโทรด/สารละลาย กับการเปลี่ยนแปลงทางเคมีที่เกิดขึ้น เครื่องมือ (ปัจจุบันรู้จักกันในชื่อแก๊สคูลอมมิเตอร์) ถูกสร้างขึ้นเพื่อวัดปริมาณไฟฟ้าจากปริมาตรของไฮโดรเจนและออกซิเจนที่ปล่อยออกมาในเซลล์อิเล็กโทรไลต์ และแสดงให้เห็น (1833) ว่าปริมาณไฟฟ้าที่ต้องใช้เพื่อให้ได้ปริมาณที่กำหนด สารไม่ได้ขึ้นอยู่กับขนาดของอิเล็กโทรด ระยะห่างระหว่างอิเล็กโทรดกับจำนวนแผ่นในแบตเตอรี่ที่ป้อนให้กับเซลล์ นอกจากนี้ ฟาราเดย์ยังค้นพบว่าปริมาณของสารที่ปล่อยออกมาระหว่างอิเล็กโทรไลซิสเป็นสัดส่วนโดยตรงกับค่าเทียบเท่าทางเคมีของสารและปริมาณไฟฟ้าที่ไหลผ่านอิเล็กโทรไลต์ (สารเคมีที่เทียบเท่าคือจำนวนกรัมของธาตุหรือสารประกอบที่ทำปฏิกิริยากับหรือแทนที่ไฮโดรเจนในสารประกอบหนึ่งโมล (1.0078 กรัม) ซม. มวลเท่ากัน) บทบัญญัติพื้นฐานทั้งสองนี้เรียกว่ากฎของฟาราเดย์ ฟาราเดย์ร่วมกับเพื่อนของเขา ดับบลิว วีเวลล์ ผู้เชี่ยวชาญด้านอักษรศาสตร์คลาสสิก ได้พัฒนาคำศัพท์ใหม่ในสาขาเคมีไฟฟ้าด้วย เขาเรียกว่าตัวนำที่แช่อยู่ในสารละลายอิเล็กโทรด (ก่อนหน้านี้เรียกว่าขั้ว) แนะนำแนวคิดของ "อิเล็กโทรไลซิส" (การเปลี่ยนแปลงทางเคมีที่เกี่ยวข้องกับการผ่านของกระแส), "อิเล็กโทรไลต์" (การนำของเหลวในเซลล์ไฟฟ้าเคมี), "แอโนด" (อิเล็กโทรดที่เกิดปฏิกิริยาออกซิเดชัน) และ "แคโทด" (อิเล็กโทรดที่ ปฏิกิริยารีดักชั่นเกิดขึ้น) เขาเรียกตัวพาประจุในไอออนของเหลว (จากภาษากรีก "คนพเนจร", "คนพเนจร") และไอออนที่เคลื่อนที่ไปทางขั้วบวก (อิเล็กโทรดบวก) ถูกเรียกว่า "แอนไอออน" และไอออนที่เคลื่อนที่ไปทางแคโทด - "ไพเพอร์" การวิจัยของฟาราเดย์เกี่ยวกับการเหนี่ยวนำแม่เหล็กไฟฟ้านำไปสู่การสร้างเครื่องกำเนิดไฟฟ้า ซึ่งทำให้สามารถดำเนินกระบวนการไฟฟ้าเคมีในระดับอุตสาหกรรมได้
ฟาราเดย์อธิบายความสามารถในการแก้ปัญหาเพื่อส่งผ่านกระแสไฟฟ้าเมื่อมีไอออนอยู่ในนั้น แต่ตัวเขาเองและนักวิทยาศาสตร์คนอื่นๆ เช่น I. Hittorf และ F. Kohlrausch เชื่อว่าไอออนจะปรากฏภายใต้อิทธิพลของกระแส ในปี 1884 เอส. อาร์เรเนียสเสนอว่า อันที่จริง ไอออนจะเกิดขึ้นได้ก็ต่อเมื่อเกลือละลายในน้ำ ผลงานของ S. Arrhenius, J. Van't Hoff และ W. Ostwald ถือเป็นเหตุการณ์สำคัญในการพัฒนาทฤษฎีอิเล็กโทรไลต์และแนวคิดเกี่ยวกับคุณสมบัติทางเคมีฟิสิกส์ของสารละลายและอุณหพลศาสตร์ของสารละลาย ความสอดคล้องกันระหว่างทฤษฎีและข้อมูลการทดลองเกี่ยวกับการนำไอออนิกและสมดุลในสารละลายมีความสมบูรณ์มากขึ้นหลังจากที่ P. Debye และ E. Hückel พิจารณาถึงปฏิกิริยาระหว่างไฟฟ้าสถิตในระยะยาวระหว่างไอออนในปี 1923
การมีส่วนร่วมที่สำคัญในอุณหพลศาสตร์เคมีไฟฟ้าและโดยเฉพาะในการอธิบายธรรมชาติของศักย์ไฟฟ้า (แรงดันไฟฟ้า) ในเซลล์ไฟฟ้าเคมีและความสมดุลระหว่างพลังงานไฟฟ้า เคมี และความร้อน จัดทำโดย J. Gibbs และ W. Nernst ศักย์ไฟฟ้าเคมีถูกกำหนดโดยพลังงานเคมีของกระบวนการที่เกิดขึ้นในเซลล์ แต่ยังขึ้นอยู่กับความเร็ว (จลนศาสตร์) ด้วย การสร้างแบบจำลองกระบวนการจลน์บนอิเล็กโทรดดำเนินการโดย Y. Tafel (1905), J. Butler (1924), M. Volmer (1930), A. N. Frumkin (1930–1933)
เซลล์ไฟฟ้าเคมี
เซลล์ไฟฟ้าเคมีมักประกอบด้วยครึ่งเซลล์สองเซลล์ ซึ่งแต่ละเซลล์เป็นอิเล็กโทรดที่จุ่มอยู่ในอิเล็กโทรไลต์ของมันเอง อิเล็กโทรดทำจากวัสดุที่เป็นสื่อกระแสไฟฟ้า (โลหะหรือคาร์บอน) หรือน้อยกว่าจากเซมิคอนดักเตอร์ ตัวพาประจุในอิเล็กโทรไลต์คืออิเล็กตรอน และตัวพาประจุในอิเล็กโทรไลต์คือไอออน สารละลายเกลือแกงที่เป็นน้ำ (โซเดียมคลอไรด์ NaCl) ซึ่งเป็นอิเล็กโทรไลต์ประกอบด้วยอนุภาคที่มีประจุ: โซเดียมไอออนบวก Na + และแอนไอออนคลอรีน Cl – หากคุณวางสารละลายดังกล่าวไว้ในสนามไฟฟ้า ไอออน Na + จะเคลื่อนที่ไปที่ขั้วลบ และ Cl – ไอออนจะเคลื่อนที่ไปยังขั้วบวก เกลือหลอมเหลว เช่น NaCl ก็เป็นอิเล็กโทรไลต์เช่นกัน อิเล็กโทรไลต์ก็สามารถเป็นของแข็งได้เช่นกัน ข- อลูมินา (โซเดียมโพลีอะลูมิเนต) ที่มีโซเดียมไอออนเคลื่อนที่หรือโพลีเมอร์แลกเปลี่ยนไอออน
ครึ่งเซลล์ถูกคั่นด้วยฉากกั้น ซึ่งไม่รบกวนการเคลื่อนที่ของไอออน แต่ป้องกันการผสมอิเล็กโทรไลต์ บทบาทของฉากกั้นดังกล่าวสามารถทำได้โดยใช้สะพานเกลือ หลอดที่มีสารละลายในน้ำปิดที่ปลายทั้งสองข้างด้วยใยแก้ว เมมเบรนแลกเปลี่ยนไอออน หรือแผ่นกระจกที่มีรูพรุน อิเล็กโทรดทั้งสองของเซลล์อิเล็กโทรไลต์สามารถจุ่มลงในอิเล็กโทรไลต์เดียวกันได้
เซลล์ไฟฟ้าเคมีมีสองประเภท: เซลล์กัลวานิกและเซลล์อิเล็กโทรไลต์ (อิเล็กโทรไลเซอร์) ในเซลล์กัลวานิก ปฏิกิริยาเคมีเกิดขึ้นเองตามธรรมชาติที่ส่วนต่อประสานของอิเล็กโทรด/อิเล็กโทรไลต์ และอิเล็กโทรดจะเชื่อมต่อถึงกันด้วยตัวนำ เซลล์กัลวานิกหลายเซลล์ที่เชื่อมต่อกันแบบอนุกรมจะสร้างแบตเตอรี่ซึ่งเป็นแหล่งจ่ายกระแสไฟฟ้าทางเคมี ในเซลล์อิเล็กโทรไลต์ ปฏิกิริยาที่ส่วนต่อประสานอิเล็กโทรด/อิเล็กโทรไลต์เกิดขึ้นเนื่องจากแหล่งพลังงานไฟฟ้าภายนอก ส่วนหลังจะถูกแปลงเป็นพลังงานเคมีของผลิตภัณฑ์ปฏิกิริยาที่เกิดขึ้นที่ขั้วไฟฟ้า โครงสร้างของเซลล์กัลวานิกแสดงไว้ในรูปที่ 1 1 และอิเล็กโทรไลเซอร์ - ในรูป. 2. โปรดทราบว่าเซลล์เดียวกันนั้นอาจทำงานเป็นเซลล์กัลวานิกหรืออิเล็กโทรไลเซอร์ก็ได้ ทั้งนี้ขึ้นอยู่กับโหมดการทำงาน ดังนั้นแบตเตอรี่รถยนต์กรดตะกั่วจึงทำหน้าที่เป็นเซลล์กัลวานิกเมื่อใช้ในการสตาร์ทเครื่องยนต์ (ขณะคายประจุ) และทำหน้าที่เป็นอิเล็กโทรไลเซอร์เมื่อชาร์จจากเครื่องกำเนิดไฟฟ้าหรือเครื่องชาร์จในรถยนต์
เซลล์กัลวานิกอย่างง่าย สร้างขึ้นในปี 1836 โดย J. Daniel (รูปที่ 1) ประกอบด้วยอิเล็กโทรด 2 อิเล็กโทรด: สังกะสี แช่อยู่ในสารละลายที่เป็นน้ำของซิงค์ซัลเฟต และทองแดง จุ่มในสารละลายน้ำของคอปเปอร์ (II) ซัลเฟต องค์ประกอบดังกล่าวคล้ายกับคู่ทองแดง-สังกะสีในคอลัมน์โวลตาอิก เมื่อวงจรภายนอกปิด อะตอมของสังกะสีบนพื้นผิวของอิเล็กโทรดสังกะสีจะถูกออกซิไดซ์เป็นไอออนโดยปล่อยอิเล็กตรอน: Zn ® Zn 2+ + 2e – อิเล็กตรอนเหล่านี้เคลื่อนที่ไปตามวงจรภายนอกไปยังอิเล็กโทรดทองแดงและลดไอออนของทองแดงให้เป็นอะตอม: Cu 2+ + 2e – ® Cu การไหลของอิเล็กตรอนในวงจรภายนอกคือกระแสที่สร้างโดยองค์ประกอบ ปฏิกิริยาโดยรวมที่นำไปสู่การเปลี่ยนแปลงทางเคมีและการสร้างพลังงานไฟฟ้ามีรูปแบบ
ปฏิกิริยาเดียวกันนี้เกิดขึ้นเมื่อเติมสังกะสีโลหะลงในสารละลายคอปเปอร์ซัลเฟต แต่ในกรณีนี้พลังงานเคมีจะถูกแปลงเป็นพลังงานความร้อน
เซลล์ไฟฟ้าเคมีมักจะแสดงเป็นแผนผังโดยแสดงขอบเขตระหว่างอิเล็กโทรดและอิเล็กโทรไลต์ด้วยแนวตั้งหรือเครื่องหมายทับ (| หรือ /) และสะพานเกลือที่มีเครื่องหมายทับสองอัน (//) ดังนั้น เซลล์กัลวานิกในรูป 1 คำตอบ
โดยที่ M คือความเข้มข้นโมลของสารละลาย
ในเซลล์อิเล็กโทรไลต์ที่แสดงในรูปที่. 2 ปฏิกิริยาเดียวกันนี้เกิดขึ้นเช่นเดียวกับในอิเล็กโทรไลเซอร์อุตสาหกรรมเพื่อผลิตคลอรีนและอัลคาไล: การเปลี่ยนน้ำเกลือ (สารละลายโซเดียมคลอไรด์เข้มข้น) เป็นคลอรีนและโซเดียมไฮดรอกไซด์ NaOH:
ไอออนคลอไรด์บนอิเล็กโทรดกราไฟต์จะถูกออกซิไดซ์เป็นก๊าซคลอรีน และน้ำบนอิเล็กโทรดเหล็กจะถูกรีดิวซ์เป็นไฮโดรเจนและไฮดรอกไซด์ไอออน อิเล็กโทรไลต์ยังคงมีความเป็นกลางทางไฟฟ้าเนื่องจากการเคลื่อนตัวของโซเดียมไอออนผ่านพาร์ติชัน - เมมเบรนแลกเปลี่ยนไอออน อิเล็กโทรดที่เกิดออกซิเดชัน (สังกะสีในรูปที่ 1 และกราไฟท์ในรูปที่ 2) เรียกว่าขั้วบวก และอิเล็กโทรดที่เกิดปฏิกิริยารีดักชันเรียกว่าแคโทด
ศักยภาพของอิเล็กโทรด
พารามิเตอร์ทางไฟฟ้าหลักของเซลล์ไฟฟ้าเคมีคือกระแส (วัดเป็นแอมแปร์, A) และศักย์ไฟฟ้า (วัดเป็นโวลต์, V) ความแรงของกระแสไฟฟ้าถูกกำหนดโดยอัตราของปฏิกิริยาอิเล็กโทรด และศักยภาพถูกกำหนดโดยพลังงานเคมีของกระบวนการที่เกิดขึ้นในเซลล์ มีค่าเท่ากับพลังงาน (วัดเป็นจูล J) หารด้วยปริมาณไฟฟ้า (วัดเป็นคูลอมบ์ C) กล่าวคือ 1 โวลต์ = 1 เจ / Cl. ดังนั้นศักยภาพขององค์ประกอบ (แรงเคลื่อนไฟฟ้า, แรงเคลื่อนไฟฟ้า) จึงเป็นการวัดพลังงานที่เกิดขึ้นระหว่างปฏิกิริยาที่เกิดขึ้น หากวงจรภายนอกเปิดอยู่ จะไม่มีปฏิกิริยาอิเล็กโทรดเกิดขึ้น
ศักยภาพของเซลล์กัลวานิกที่มีวงจรภายนอกแบบเปิดจะให้ข้อมูลเกี่ยวกับอุณหพลศาสตร์ของปฏิกิริยาของมัน ศักยภาพขององค์ประกอบที่แสดงในรูป 1 ที่ความเข้มข้นของสารละลาย 1 M และอุณหภูมิ 25 ° C - ศักย์มาตรฐาน อี° – เท่ากับ 1.10 V. พลังงานที่สอดคล้องกัน, ศักย์ทางอุณหพลศาสตร์ของ Gibbs, D ช°มอบให้โดย
ที่ไหน n– จำนวนอิเล็กตรอนที่ถูกถ่ายโอนระหว่างปฏิกิริยา (ในกรณีนี้คือ 2) เอฟ– หมายเลขฟาราเดย์ (96 485 C / ตุ่น). ศักยภาพของเซลล์กัลวานิกเท่ากับความต่างศักย์ระหว่างครึ่งเซลล์ทั้งสองของมัน นั่นคือ ความต่างศักย์ไฟฟ้าของอิเล็กโทรด ศักย์ไฟฟ้าของอิเล็กโทรดจะถูกวัดโดยสัมพันธ์กับศักย์ไฟฟ้าของอิเล็กโทรดอ้างอิง ซึ่งโดยทั่วไปแล้วจะกำหนดให้เป็นศูนย์ ( ซม. โต๊ะ). ตามข้อตกลง อิเล็กโทรดไฮโดรเจนปกติ (N.H.E.) ได้รับเลือกเป็นอิเล็กโทรดมาตรฐาน เป็นแผ่นแพลตตินัมซึ่งเคลือบด้วยแพลตตินัมแบล็คอิ่มตัวด้วยก๊าซไฮโดรเจนที่ความดัน 1.01H 10 5 Pa (1 atm.) และแช่ในสารละลายที่มี H + ไอออนด้วยกิจกรรมทางอุณหพลศาสตร์ ก= 1 ตามแผนผัง อิเล็กโทรดนี้สามารถแสดงเป็น Pt/H 2 (1.01H 10 5 Pa)/H + ( ก= 1) ปฏิกิริยา 2H + + 2e – ® H 2 เกิดขึ้นกับมัน ในการกำหนดศักย์ไฟฟ้ามาตรฐานของอิเล็กโทรดทองแดง Cu/Cu 2+ ให้ประกอบเซลล์กัลวานิกต่อไปนี้:
และสำหรับครึ่งปฏิกิริยา Cu 2+ + 2e – ® Cu การวัดจะให้
ในทำนองเดียวกัน สำหรับครึ่งเซลล์ Zn/Zn 2+ ซึ่งเกิดปฏิกิริยา Zn 2+ + 2e – ® Zn เราจะได้
ความแตกต่างระหว่างศักย์ไฟฟ้ามาตรฐานทั้งสองนี้เท่ากับศักย์ไฟฟ้ามาตรฐานขององค์ประกอบ Zn–Cu
อันที่จริง อิเล็กโทรดไฮโดรเจนไม่ค่อยถูกนำมาใช้ในการวัดค่าโพเทนชิโอเมตริก เนื่องจากอิเล็กโทรดนี้เป็นระบบในอุดมคติที่ยากต่อการนำไปใช้ในทางปฏิบัติ บ่อยครั้งที่มีการใช้อิเล็กโทรดเปรียบเทียบประเภทต่างๆ ที่สะดวกกว่าและกะทัดรัดกว่า โดยมีค่าศักยภาพที่วัดได้เฉพาะเจาะจงและระมัดระวังโดยสัมพันธ์กับกระแสไฟฟ้า โดยปกติแล้วจะใช้อิเล็กโทรดคาโลเมล (CE) ซึ่งประกอบด้วยปรอทโลหะ ปรอทคลอไรด์ (คาโลเมล) และสารละลายโพแทสเซียมคลอไรด์: Hg /Hg 2 Cl 2 /KCl ปฏิกิริยาต่อไปนี้เกิดขึ้นที่อิเล็กโทรด:
ศักยภาพเคอี ขึ้นอยู่กับความเข้มข้นของไอออนปรอท และอย่างหลังขึ้นอยู่กับความเข้มข้นของสารละลาย KCl สำหรับสารละลาย KCl ที่อิ่มตัว อี° (ke.) av = 0.2412 V (n.e.) ที่ 25° C
|
ศักยภาพอิเล็กโทรดมาตรฐาน |
|
| ปฏิกิริยาอิเล็กโทรด |
อีโอ วี |
| หลี่ + + อี – ® หลี่ | |
| มก. 2+ + 2e – ® มก | |
| อัล 3+ + 3e – ® อัล | |
| สังกะสี 2+ + 2e – ® สังกะสี | |
| Cr 3+ + อี – ® Cr 2+ | |
| 2H + + 2e – ® H 2 | |
| ลูกบาศ์ก 2+ + 2e – ® ลูกบาศ์ก | |
| เฟ 3+ + อี – ® เฟ 2+ | |
| O 2 + 4H + + 4e – ® 2H 2 O | |
| Cl 2 + 2e – ® 2Cl – | |
| ฉ 2 + 2e – ® 2F – | |
โปรดทราบว่าสสารของอิเล็กโทรดบางชนิดไม่รวมอยู่ในสมการของปฏิกิริยาที่เกี่ยวข้อง ใช่ปฏิกิริยา
เกิดขึ้นจริงบนอิเล็กโทรดแพลทินัมในเซลล์ Pt/Fe 3+, Fe 2+ อิเล็กโทรดแพลทินัมเป็นแบบเฉื่อยและให้การสัมผัสกับอิเล็กโทรไลต์ที่มีองค์ประกอบในรูปแบบออกซิไดซ์และรีดิวซ์เท่านั้น (ในกรณีนี้คือไอออนของเหล็กไดวาเลนต์และไตรวาเลนต์) อิเล็กโทรดแพลทินัมมีบทบาทเหมือนกันใน N.V.E.
ตารางศักย์ไฟฟ้าของอิเล็กโทรดทำให้คุณสามารถคำนวณ EMF ของเซลล์กัลวานิกโดยพิจารณาจากศักย์ไฟฟ้าของอิเล็กโทรด นอกจากนี้ยังสามารถใช้เพื่อทำนายว่าปฏิกิริยารีดอกซ์จะเกิดขึ้นหรือไม่ เราสามารถพูดถึงศักย์อิเล็กโทรดมาตรฐานได้ก็ต่อเมื่อกิจกรรมของส่วนประกอบที่เข้าร่วมในปฏิกิริยาเท่ากับ 1 นั่นคือ ความเข้มข้นในสารละลายอยู่ใกล้ 1M ศักยภาพของอิเล็กโทรด อีขึ้นอยู่กับความเข้มข้นของรูปแบบออกซิไดซ์และรูปแบบรีดิวซ์ในสารละลาย และสัมพันธ์กับสิ่งเหล่านั้นและศักยภาพมาตรฐาน อี°สมการเนิร์สต์ สำหรับปฏิกิริยาทั่วไป
วัว + nอี – = สีแดง
สมการนี้มีรูปแบบ
ที่ไหน ร– ค่าคงที่ก๊าซสากล ต– อุณหภูมิสัมบูรณ์ และ – กิจกรรมของรูปแบบออกซิไดซ์และรูปรีดิวซ์ กิจกรรมของของแข็งและของเหลวบริสุทธิ์ถือเป็น 1 ที่ 25° C RT/F= 0.025 V โดยการวัดศักย์ไฟฟ้าของอิเล็กโทรดสัมพันธ์กับศักย์ของอิเล็กโทรดอ้างอิง จึงสามารถระบุความเข้มข้นของสารในสารละลายได้โดยใช้สมการ (10) วิธีนี้เรียกว่าโพเทนชิโอเมทรี
ปฏิกิริยาอิเล็กโทรด
การวัดโพเทนชิโอเมตริกจะดำเนินการภายใต้สภาวะเมื่อไม่มีกระแสในเซลล์ไฟฟ้าเคมี ซึ่งหมายความว่าไม่มีการเปลี่ยนแปลงทางเคมีโดยรวมเกิดขึ้น และศักยภาพที่วัดได้ (สมดุล) จะถูกกำหนดโดยอุณหพลศาสตร์ของปฏิกิริยา ภายใต้สภาวะเหล่านี้ ปัจจัยต่างๆ เช่น ขนาดและรูปร่างของอิเล็กโทรดหรือความเข้มของการกวนสารละลาย จะไม่ส่งผลต่อศักยภาพที่วัดได้ หากกระแสไหลผ่านเซลล์ไฟฟ้าเคมี อัตราของปฏิกิริยาอิเล็กโทรดไม่เพียงขึ้นอยู่กับพารามิเตอร์ทางอุณหพลศาสตร์เท่านั้น แต่ยังขึ้นอยู่กับความแรงของกระแสตามสมการด้วย
ที่ไหน n– จำนวนอิเล็กตรอนที่มีส่วนร่วมในปฏิกิริยาอิเล็กโทรดที่กำหนด เอฟ– หมายเลขฟาราเดย์ ในกรณีนี้ ศักยภาพของเซลล์ไฟฟ้าเคมีขึ้นอยู่กับปัจจัยจลน์ เช่นเดียวกับวัสดุที่ใช้สร้างอิเล็กโทรด ขนาดและรูปร่างของอิเล็กโทรด ความเข้มของการกวนสารละลาย และปัจจัยอื่นๆ อีกมากมาย ความต้านทานภายในของเซลล์ไม่สามารถละเลยได้ นอกจากความต่างศักย์ที่ขอบเขตทั้งอิเล็กโทรด/อิเล็กโทรไลต์แล้ว แรงดันตกคร่อมยังเกิดขึ้นในสารละลายด้วย เนื่องจากความต้านทาน แรงดันไฟฟ้าตกทำให้ยากต่อการศึกษาผลกระทบที่เกี่ยวข้องกับปฏิกิริยาที่เกิดขึ้นที่ขั้วไฟฟ้าทั้งสอง โดยปกติแล้วจะทำการศึกษาปฏิกิริยาบนอิเล็กโทรดตัวเดียวซึ่งเรียกว่าอิเล็กโทรดทำงานหรือตัวบ่งชี้โดยใช้เซลล์สามอิเล็กโทรด (รูปที่ 3): อิเล็กโทรดที่สาม (เช่นคาโลเมลอิ่มตัว) จะถูกวางไว้ในช่องเดียวกับการทำงาน ประการแรกให้อยู่ใกล้ที่สุดเท่าที่จะเป็นไปได้เพื่อลดผลกระทบจากแรงดันตกคร่อมโอห์มมิกให้เหลือน้อยที่สุด โดยการวัดกระแสผ่านอิเล็กโทรดทำงานเป็นฟังก์ชันของศักย์ไฟฟ้าของอิเล็กโทรดนี้สัมพันธ์กับอิเล็กโทรดอ้างอิงที่เรียกว่า เส้นโค้งโพลาไรเซชัน
เมื่อกระแสไฟฟ้าภายนอกผ่านไป ศักย์ไฟฟ้าจะแตกต่างจากสมดุล การเบี่ยงเบนนี้เรียกว่าโพลาไรเซชัน และขนาดของมันเรียกว่าแรงดันไฟฟ้าเกิน แรงดันไฟฟ้าเกินขึ้นอยู่กับปัจจัยหลายประการที่จำกัดอัตราปฏิกิริยาของอิเล็กโทรด ปฏิกิริยาอิเล็กโทรดเร็วที่ความหนาแน่นกระแสที่กำหนด (ความแรงของกระแสต่อหน่วยพื้นผิวอิเล็กโทรด) เกิดขึ้นที่ศักย์ไฟฟ้าใกล้กับเทอร์โมไดนามิกส์ และด้วยเหตุนี้ที่แรงดันไฟฟ้าเกินต่ำ ปฏิกิริยาที่ช้านั้นมีลักษณะเฉพาะคือแรงดันไฟฟ้าเกินสูง อัตราของปฏิกิริยาอิเล็กโทรดและด้วยเหตุนี้แรงดันไฟฟ้าเกินจึงขึ้นอยู่กับความเข้มข้นของรีเอเจนต์ อุณหภูมิ ตัวทำละลาย วัสดุอิเล็กโทรด วิธีและความเร็วของการถ่ายโอนมวล และความหนาแน่นกระแส แรงดันไฟฟ้าเกินทั้งหมดสามารถแบ่งออกเป็นหลายองค์ประกอบ: ความเข้มข้น การกระตุ้น และปฏิกิริยา
ความเข้มข้นของแรงดันไฟฟ้าเกินเกิดจากข้อเท็จจริงที่ว่าเมื่อกระแสไฟฟ้าไหลผ่าน ความเข้มข้นของไอออนที่ทำปฏิกิริยาบนพื้นผิวของอิเล็กโทรดจะเปลี่ยนไป เนื่องจากบริเวณนี้ใช้สารที่ออกฤทธิ์ทางไฟฟ้าและผลิตภัณฑ์ที่เกิดปฏิกิริยา ให้เราพิจารณาการลดลงของ Cu 2+ บนอิเล็กโทรดทองแดง เริ่มแรกความเข้มข้นของ Cu 2+ ในสารละลายคือ 1M ในกรณีที่ไม่มีกระแสไฟฟ้าในวงจร ศักย์ไฟฟ้าของอิเล็กโทรดทองแดงจะใกล้เคียงกับศักย์มาตรฐานของคู่ Cu/Cu 2+ กล่าวคือ 0.34 V สัมพันธ์กับ N.E. [ ซม. สมการ (6)] เมื่อกระแสแคโทดผ่านไป ความเข้มข้นของไอออน Cu 2+ บนพื้นผิวอิเล็กโทรดจะลดลง และศักยภาพของแคโทดตามสมการ Nernst (10) จะกลายเป็นลบมากขึ้น ส่วนใหม่ของรีเอเจนต์จะถูกส่งจากสารละลายไปยังอิเล็กโทรดด้วยวิธีต่างๆ กัน ซึ่งเป็นผลมาจากการแพร่กระจาย การพาความร้อน และการย้ายถิ่น ยิ่งความเร็วของกระบวนการเหล่านี้สูง (เช่น การผสมยิ่งเข้มข้นมากขึ้น) ความเข้มข้นของแรงดันไฟฟ้าเกินก็จะยิ่งลดลง ส่วนประกอบนี้ของแรงดันไฟฟ้าเกินรวมนี้สามารถคำนวณได้บ่อยครั้ง หากโพลาไรเซชันของความเข้มข้นมีส่วนสำคัญต่อแรงดันไฟฟ้าเกินทั้งหมด (ซึ่งหมายความว่าอัตราของขั้นตอนที่เหลือของปฏิกิริยาอิเล็กโทรดนั้นสูง) ปฏิกิริยาดังกล่าวจะเรียกว่าปฏิกิริยาแบบย้อนกลับได้หรือแบบเนิร์นสเตียน
แรงดันไฟฟ้าเกินในการเปิดใช้งานเกิดขึ้นเนื่องจากการถ่ายโอนอิเล็กตรอนบนพื้นผิวอิเล็กโทรดไม่ได้เกิดขึ้นทันที แต่เกิดขึ้นที่ความเร็วจำกัด ให้เราพิจารณาปฏิกิริยาอิเล็กโทรดทั่วไป ox + nอี – = สีแดง ในการถ่ายโอนอิเล็กตรอนไปยังสารประกอบออกซิไดซ์ในอัตราที่กำหนด (เช่น ที่ความหนาแน่นกระแสที่กำหนด) จำเป็นต้องเอาชนะอุปสรรคพลังงานที่เรียกว่าพลังงานกระตุ้นของปฏิกิริยาอิเล็กโทรด พลังงานนี้มาจากศักยภาพที่ใช้ ความสัมพันธ์ระหว่างความหนาแน่นกระแสและแรงดันไฟฟ้าเกินกระตุ้นอธิบายไว้ในสมการของบัตเลอร์–โวลเมอร์และทาเฟล ซึ่งสามารถใช้เพื่อกำหนดพารามิเตอร์จลน์ของปฏิกิริยาอิเล็กโทรด ปฏิกิริยาอิเล็กโทรดหลายอย่าง เช่น ปฏิกิริยารีดักชันของน้ำเป็นไฮโดรเจน และปฏิกิริยาออกซิเดชันของน้ำเป็นออกซิเจน เกิดขึ้นอย่างช้าๆ อัตราการเกิดปฏิกิริยาของอิเล็กโทรดอาจขึ้นอยู่กับวัสดุที่ใช้สร้างอิเล็กโทรดและคุณสมบัติของพื้นผิวเป็นอย่างมาก ตัวอย่างเช่น บนอิเล็กโทรดปรอท การลดน้ำให้เป็นไฮโดรเจนเป็นเรื่องยากอย่างมาก โดยมีลักษณะเฉพาะคือแรงดันไฮโดรเจนสูงเกินไป ปฏิกิริยานี้เกิดขึ้นเร็วกว่ามากและมีแรงดันไฟฟ้าเกินบนแพลตตินัมน้อยกว่า ด้วยเหตุนี้จึงมีการใช้แพลตตินัมในอิเล็กโทรดอ้างอิงไฮโดรเจน อัตราการเกิดปฏิกิริยายังขึ้นอยู่กับสารที่ถูกดูดซับหรือจับกับพื้นผิวอิเล็กโทรด ดังนั้นไซยาไนด์ไอออนและสารประกอบอินทรีย์จำนวนหนึ่งจึงลดอัตราการวิวัฒนาการของไฮโดรเจนบนพื้นผิวของอิเล็กโทรดแพลทินัม ในเวลาเดียวกันสารลดแรงตึงผิวบางชนิดสามารถเพิ่มอัตราการเกิดปฏิกิริยาของอิเล็กโทรดได้อย่างมาก - เรียกว่าอิเล็กโทรคะตะลิสต์
แรงดันไฟเกินจากปฏิกิริยาเกิดขึ้นเมื่อการถ่ายโอนอิเล็กตรอนที่อิเล็กโทรดควบคู่ไปกับปฏิกิริยาเคมีในสารละลาย ปฏิกิริยาดังกล่าวสามารถทำหน้าที่เป็นแหล่งกำเนิดของอนุภาคที่เกี่ยวข้องกับการถ่ายโอนอิเล็กตรอน และในขณะเดียวกันก็จำกัดอัตราของกระบวนการอิเล็กโทรดทั้งหมด ด้วยเหตุนี้การทราบรายละเอียดของกลไก (เช่น ระยะและสถานะระหว่างกลาง) ของปฏิกิริยาอิเล็กโทรดจึงเป็นสิ่งสำคัญมาก ในหลายกรณี วัสดุตั้งต้นต้องผ่านการเปลี่ยนแปลงหลายครั้งก่อนที่จะกลายเป็นผลิตภัณฑ์ขั้นสุดท้ายที่อิเล็กโทรด เช่น ในกรณีของการลดออกซิเจนลงสู่น้ำ ซึ่งเป็นกระบวนการที่มีความสำคัญในทางปฏิบัติอย่างยิ่ง ปฏิกิริยารวมจะมีรูปแบบ
และประกอบด้วยหลายขั้นตอน โดยขั้นตอนหนึ่งคือพันธะออกซิเจนและออกซิเจนที่ถูกทำลาย เนื่องจากปฏิกิริยาหลายขั้นตอนนี้ ปฏิกิริยาที่อิเล็กโทรดส่วนใหญ่จะช้า และในระดับอุตสาหกรรมจะดำเนินการต่อหน้าตัวเร่งปฏิกิริยาด้วยไฟฟ้า มีการศึกษากลไกของปฏิกิริยาอิเล็กโทรดโดยใช้วิธีการวิเคราะห์ทางไฟฟ้าที่อธิบายไว้ด้านล่าง บ่อยครั้งที่ปฏิกิริยาเกิดการเปลี่ยนแปลงเมื่อองค์ประกอบของสารละลายและลักษณะของตัวทำละลายเปลี่ยนไป ตัวอย่างเช่น การลดลงของออกซิเจนในอะซิโตไนไตรล์ ซึ่งมีโปรตอนขาด จะดำเนินการตามกลไกอิเล็กตรอนเดี่ยวอย่างง่าย:
วิธีวิเคราะห์เคมีไฟฟ้า
วิธีการเคมีไฟฟ้าหลายชนิดได้รับการพัฒนาสำหรับการวิเคราะห์ทางเคมีในเชิงคุณภาพและเชิงปริมาณ ซึ่งมักจะมีประโยชน์ในการกำหนดพารามิเตอร์ทางอุณหพลศาสตร์และจลน์ของปฏิกิริยาอิเล็กโทรดและศึกษากลไกของปฏิกิริยาด้วย
การนำไฟฟ้า
ขึ้นอยู่กับการวัดค่าการนำไฟฟ้าของสารละลายและใช้ในการกำหนดความเข้มข้นของเกลือ กรด เบส ฯลฯ ในการวัดค่าสื่อไฟฟ้า โดยปกติจะใช้อิเล็กโทรดที่ทำจากวัสดุที่เหมือนกัน และเลือกเงื่อนไขสำหรับการดำเนินการในลักษณะที่จะลดการกระโดดที่อาจเกิดขึ้นที่ทั้งส่วนต่อประสานอิเล็กโทรด/อิเล็กโทรไลต์ (เช่น ใช้กระแสสลับความถี่สูง ). ในกรณีนี้ ปัจจัยหลักต่อศักย์ไฟฟ้าของเซลล์ที่วัดได้นั้นเกิดจากแรงดันตกคร่อมโอห์มมิก นักลงทุนสัมพันธ์, ที่ไหน ร– ความต้านทานต่อสารละลาย ค่าการนำไฟฟ้าของสารละลายที่มีองค์ประกอบเดียวสามารถสัมพันธ์กับความเข้มข้นได้ และการวัดค่าการนำไฟฟ้าของอิเล็กโทรไลต์ที่มีองค์ประกอบเชิงซ้อนทำให้สามารถประมาณปริมาณไอออนทั้งหมดในสารละลาย และนำไปใช้ เช่น ในการตรวจสอบคุณภาพของการกลั่น หรือน้ำปราศจากไอออน ในการวัดค่าการนำไฟฟ้าอีกประเภทหนึ่ง ซึ่งก็คือ การไตเตรทแบบการนำไฟฟ้า จะมีการเพิ่มรีเอเจนต์ที่ทราบไว้ในส่วนต่างๆ ของสารละลายที่วิเคราะห์ และติดตามการเปลี่ยนแปลงของการนำไฟฟ้า จุดสมมูลซึ่งมีการบันทึกการเปลี่ยนแปลงการนำไฟฟ้าอย่างกะทันหันถูกกำหนดจากกราฟของการพึ่งพาค่านี้กับปริมาตรของรีเอเจนต์ที่เพิ่ม
โพเทนชิโอเมทรี
ใช้เพื่อกำหนดพารามิเตอร์เคมีกายภาพต่างๆ ตามข้อมูลศักยภาพของเซลล์กัลวานิก ศักย์ไฟฟ้าในกรณีที่ไม่มีกระแสในวงจรไฟฟ้าเคมี ซึ่งวัดสัมพันธ์กับอิเล็กโทรดอ้างอิง มีความสัมพันธ์กับความเข้มข้นของสารละลายโดยสมการ Nernst ในการวัดค่าโพเทนชิโอเมตริก อิเล็กโทรดแบบเจาะจงไอออนถูกนำมาใช้กันอย่างแพร่หลายซึ่งมีความไวต่อไอออน 1 ตัวในสารละลายเป็นหลัก ได้แก่ อิเล็กโทรดแก้วสำหรับวัดค่า pH และอิเล็กโทรดสำหรับการตรวจวัดแบบเลือกไอออนของโซเดียม แอมโมเนียม ฟลูออรีน แคลเซียม แมกนีเซียม ฯลฯ พื้นผิว ชั้นของอิเล็กโทรดคัดเลือกไอออนอาจมีเอนไซม์อยู่ด้วย และผลลัพธ์ก็คือระบบที่มีความไวต่อสารตั้งต้นที่เหมาะสม โปรดทราบว่าศักยภาพของอิเล็กโทรดคัดเลือกไอออนไม่ได้ถูกกำหนดโดยการถ่ายโอนอิเล็กตรอน เช่น ในกรณีของสารที่มีค่าการนำไฟฟ้าแบบอิเล็กทรอนิกส์ แต่โดยการถ่ายโอนหรือการแลกเปลี่ยนไอออนเป็นหลัก อย่างไรก็ตาม สมการ Nernst ซึ่งสัมพันธ์กับศักย์ไฟฟ้าของอิเล็กโทรดกับลอการิทึมของความเข้มข้น (หรือกิจกรรม) ของสารในสารละลาย ก็ใช้กับอิเล็กโทรดดังกล่าวได้เช่นกัน ในการไตเตรทแบบโพเทนชิโอเมตริก รีเอเจนต์จะถูกเติมลงในสารละลายที่กำลังวิเคราะห์เป็นบางส่วน และติดตามการเปลี่ยนแปลงในศักยภาพ คุณลักษณะเส้นโค้งรูปตัว S ของการไทเทรตประเภทนี้ช่วยให้สามารถระบุจุดสมมูลและค้นหาพารามิเตอร์ทางอุณหพลศาสตร์ เช่น ค่าคงที่สมดุลและศักย์มาตรฐาน
โวลแทมเมทรี
วิธีการโวลแทมเมทริกทุกประเภทใช้ไมโครอิเล็กโทรดที่ทำงานโดยมีพื้นที่ผิว 10–7–10–1 cm2 เส้นโค้งแรงดันไฟฟ้าปัจจุบันที่ได้รับช่วยให้สามารถระบุสารที่ละลาย กำหนดความเข้มข้นของสารเหล่านั้น และมักจะเป็นพารามิเตอร์ทางอุณหพลศาสตร์และจลน์ศาสตร์ วิธีโวลแทมเมทริกวิธีแรก - โพลาโรกราฟี - ถูกเสนอในปี พ.ศ. 2465 โดยนักเคมีชาวเช็ก เจ. เฮย์รอฟสกี้ อิเล็กโทรดที่ใช้งานอยู่ในการตั้งค่าของเขาคืออิเล็กโทรดปรอทแบบหยด ปรอทมีแรงดันไฟฟ้าเกินของไฮโดรเจนสูง ดังนั้นอิเล็กโทรดปรอทจึงสะดวกสำหรับการศึกษากระบวนการที่เกิดขึ้นที่ศักย์ไฟฟ้าลบ พื้นผิวอิเล็กโทรดได้รับการต่ออายุอย่างต่อเนื่องในระหว่างกระบวนการตรวจวัด ซึ่งช่วยลดการปนเปื้อนของอิเล็กโทรด การศึกษาโวลแทมเมตริกยังดำเนินการโดยใช้อิเล็กโทรดที่เป็นของแข็ง เช่น แพลทินัมและคาร์บอน และใช้กระบวนการที่เกิดขึ้นที่ศักย์ไฟฟ้าเชิงบวก ในโวลแทมเมทรีแบบกวาดเชิงเส้น (chronoamperometry) ศักย์ไฟฟ้าจะเปลี่ยนแปลงเป็นเส้นตรงตามเวลาและสารละลายจะไม่ถูกกวน ดังนั้นการถ่ายโอนมวลจึงเกิดขึ้นเนื่องจากการแพร่กระจายเพียงอย่างเดียว ในโวลแทมเมทรีแบบไซคลิก พัลส์แรงดันไฟฟ้ารูปสามเหลี่ยมซ้ำๆ จะถูกส่งไปยังอิเล็กโทรด สารที่เกิดขึ้นในส่วนจากน้อยไปหามากของวัฏจักรจะได้รับการศึกษาในส่วนจากมากไปหาน้อย วิธีนี้มีประสิทธิภาพโดยเฉพาะอย่างยิ่งสำหรับการศึกษากลไกของปฏิกิริยาอิเล็กโทรดโดยการวิเคราะห์เส้นโค้งโพลาไรเซชันที่อัตราการกวาดที่เป็นไปได้และความเข้มข้นของสารละลายที่แตกต่างกัน มีโวลแทมเมทรีประเภทอื่น - ดิฟเฟอเรนเชียลพัลส์และคลื่นสี่เหลี่ยม - ซึ่งพัลส์แรงดันไฟฟ้าที่มีรูปร่างต่างกันจะซ้อนทับบนศักยภาพที่เพิ่มขึ้นเชิงเส้น วิธีการเหล่านี้ใช้กันอย่างแพร่หลายเพื่อกำหนดความเข้มข้นเล็กน้อยของสารในสารละลาย หากในระหว่างการวัดโวลแทมเมทรี มีการผสมสารละลาย ซึ่งหมายความว่าการถ่ายโอนมวลเกิดขึ้นพร้อมๆ กันโดยใช้การพาความร้อนและการแพร่ เราก็พูดถึงโวลแทมเมทรีแบบอุทกพลศาสตร์ ในกรณีนี้จะสะดวกที่จะใช้อิเล็กโทรดดิสก์แบบหมุนได้เนื่องจากเส้นโค้งแรงดันไฟฟ้าปัจจุบันที่ทดลองสามารถเปรียบเทียบได้โดยตรงกับเส้นโค้งทางทฤษฎี
แอมเพอโรเมทรี
วิธีการนี้ขึ้นอยู่กับการวัดกระแสจำกัดการแพร่กระจายที่ไหลผ่านสารละลายที่แรงดันไฟฟ้าคงที่ระหว่างอิเล็กโทรดตัวบ่งชี้และอิเล็กโทรดอ้างอิง ในการไตเตรทแบบแอมเพอโรเมตริก จุดสมมูลจะถูกกำหนดโดยการแตกของกราฟปัจจุบัน ซึ่งเป็นปริมาตรของสารละลายการทำงานที่เพิ่มเข้าไป วิธีโครโนแอมเพอโรเมตริกขึ้นอยู่กับการวัดการขึ้นต่อกันของกระแสตรงเวลา และส่วนใหญ่จะใช้เพื่อกำหนดค่าสัมประสิทธิ์การแพร่และค่าคงที่ของอัตรา เซลล์ไฟฟ้าเคมีขนาดเล็กที่ทำหน้าที่เป็นเซ็นเซอร์ที่เอาต์พุตของคอลัมน์โครมาโตกราฟีของเหลวทำงานบนหลักการของแอมเพอโรเมทรี (เช่นเดียวกับโวลแทมเมทรี) วิธีการกัลวาโนสแตติกนั้นคล้ายคลึงกับวิธีแอมเพอโรเมตริก แต่จะวัดศักยภาพเมื่อมีกระแสไฟฟ้าจำนวนหนึ่งไหลผ่านเซลล์ ดังนั้นในโครโนโพเทนชิโอเมทรี การเปลี่ยนแปลงศักย์ไฟฟ้าในช่วงเวลาหนึ่งจึงถูกควบคุม วิธีการเหล่านี้ใช้เพื่อศึกษาจลนศาสตร์ของปฏิกิริยาอิเล็กโทรดเป็นหลัก
คูลอมเมตริก
ในคูลอมเมตริก ด้วยศักยภาพที่ควบคุมได้ อิเล็กโทรไลซิสที่สมบูรณ์ของสารละลายจะดำเนินการโดยการผสมอย่างเข้มข้นในอิเล็กโทรไลเซอร์ที่มีอิเล็กโทรดทำงานที่ค่อนข้างใหญ่ (ด้านล่างของปรอทหรือตาข่ายแพลตตินัม) ปริมาณไฟฟ้าทั้งหมด ( ถาม, C) ที่จำเป็นสำหรับอิเล็กโทรไลซิสสัมพันธ์กับปริมาณของสารที่ขึ้นรูป ( ก, ง) กฎของฟาราเดย์:
ที่ไหน ม- พวกเขาพูด มวล (กรัม/โมล) เอฟ- หมายเลขฟาราเดย์ การไตเตรทแบบคูลอมเมตริกเกี่ยวข้องกับการใช้กระแสคงที่เพื่อสร้างรีเอเจนต์ทางไฟฟ้าซึ่งทำปฏิกิริยากับสารที่กำลังหาค่า ความคืบหน้าของการไทเทรตจะถูกควบคุมแบบโพเทนชิโอเมตริกหรือแบบแอมเพอโรเมตริก วิธีคูลอมเมตริกนั้นสะดวกเนื่องจากมีลักษณะเป็นสัมบูรณ์ (เช่น ทำให้คุณสามารถคำนวณปริมาณของสารที่วิเคราะห์โดยไม่ต้องใช้เส้นโค้งการสอบเทียบ) และไม่ไวต่อการเปลี่ยนแปลงในสภาวะอิเล็กโทรไลซิสและพารามิเตอร์ของอิเล็กโทรไลเซอร์ (พื้นที่ผิวของอิเล็กโทรดหรือความเข้มของการกวน) ในคูลโลกราวิเมทรี ปริมาณของสารที่ผ่านการอิเล็กโทรไลซิสจะถูกกำหนดโดยการชั่งน้ำหนักอิเล็กโทรดก่อนและหลังอิเล็กโทรไลซิส
มีวิธีการวิเคราะห์ทางไฟฟ้าอื่นๆ ในโพลาโรกราฟีกระแสสลับ แรงดันไฟฟ้าไซนัสซอยด์แอมพลิจูดต่ำถูกนำไปใช้กับศักย์ไฟฟ้าที่แปรผันเชิงเส้นในช่วงความถี่กว้าง และทั้งแอมพลิจูดและการเปลี่ยนเฟสของกระแสสลับที่เป็นผลลัพธ์หรืออิมพีแดนซ์จะถูกกำหนด จากข้อมูลเหล่านี้ จะได้รับข้อมูลเกี่ยวกับธรรมชาติของสารในสารละลาย ตลอดจนกลไกและจลนศาสตร์ของปฏิกิริยาอิเล็กโทรด วิธีการแบบชั้นบางใช้เซลล์ไฟฟ้าเคมีที่มีชั้นอิเล็กโทรไลต์หนา 10–100 µm ในเซลล์ดังกล่าว อิเล็กโทรไลซิสจะดำเนินการได้เร็วกว่าอิเล็กโทรไลเซอร์ทั่วไป เพื่อศึกษากระบวนการอิเล็กโทรด จะใช้วิธีการทางสเปกโตรเคมีที่มีการลงทะเบียนสเปกโตรโฟโตเมตริก ในการวิเคราะห์สารที่เกิดขึ้นบนพื้นผิวของอิเล็กโทรด จะมีการวัดการดูดกลืนแสงในบริเวณที่มองเห็นได้ รวมถึงบริเวณ UV และ IR การเปลี่ยนแปลงคุณสมบัติของพื้นผิวอิเล็กโทรดและตัวกลางจะถูกตรวจสอบโดยใช้วิธีการสะท้อนกลับทางไฟฟ้าและวงรี ซึ่งอิงจากการวัดการสะท้อนของรังสีจากพื้นผิวอิเล็กโทรด ซึ่งรวมถึงวิธีการสะท้อนด้วยแสงและการกระเจิงของแสงรามัน (รามันสเปกโทรสโกปี) สเปกโทรสโกปีฮาร์มอนิกที่สอง (ฟูริเยร์สเปกโทรสโกปี)
ปรากฏการณ์และวิธีการเคมีไฟฟ้าอื่นๆ
ด้วยการเคลื่อนที่สัมพัทธ์ของอิเล็กโทรไลต์และอนุภาคหรือพื้นผิวที่มีประจุ ผลกระทบทางไฟฟ้าจลน์จะเกิดขึ้น ตัวอย่างที่สำคัญของประเภทนี้คืออิเล็กโทรโฟเรซิส ซึ่งเกิดการแยกอนุภาคที่มีประจุ (เช่น โมเลกุลโปรตีนหรืออนุภาคคอลลอยด์) ที่เคลื่อนที่ในสนามไฟฟ้าเกิดขึ้น วิธีการอิเล็กโทรโฟเรติกใช้กันอย่างแพร่หลายเพื่อแยกโปรตีนหรือกรดดีออกซีไรโบนิวคลีอิก (DNA) ออกเป็นเจล ปรากฏการณ์ทางไฟฟ้ามีบทบาทสำคัญในการทำงานของสิ่งมีชีวิต: มีหน้าที่ในการสร้างและการแพร่กระจายของแรงกระตุ้นเส้นประสาท, การเกิดขึ้นของศักยภาพของเมมเบรน ฯลฯ ใช้วิธีการเคมีไฟฟ้าหลายวิธีเพื่อศึกษาระบบทางชีววิทยาและส่วนประกอบต่างๆ เป็นเรื่องที่น่าสนใจที่จะศึกษาผลกระทบของแสงต่อกระบวนการไฟฟ้าเคมี ดังนั้นหัวข้อของการวิจัยโฟโตอิเล็กโตรเคมีคือการสร้างพลังงานไฟฟ้าและการเริ่มต้นปฏิกิริยาเคมีภายใต้อิทธิพลของแสง ซึ่งมีความสำคัญมากในการเพิ่มประสิทธิภาพในการแปลงพลังงานแสงอาทิตย์เป็นพลังงานไฟฟ้า อิเล็กโทรดเซมิคอนดักเตอร์ที่ทำจากไททาเนียมไดออกไซด์ แคดเมียมซัลไฟด์ แกลเลียมอาร์เซไนด์ และซิลิคอน มักใช้ที่นี่ ปรากฏการณ์ที่น่าสนใจอีกประการหนึ่งคืออิเล็กโตรเคมิลูมิเนสเซนซ์ กล่าวคือ การสร้างแสงในเซลล์ไฟฟ้าเคมี จะสังเกตได้เมื่อมีผลิตภัณฑ์พลังงานสูงเกิดขึ้นบนอิเล็กโทรด บ่อยครั้งกระบวนการนี้ดำเนินการในลักษณะวงจรเพื่อให้ได้สารประกอบที่กำหนดทั้งในรูปแบบออกซิไดซ์และรีดิวซ์ ปฏิสัมพันธ์ระหว่างกันนำไปสู่การก่อตัวของโมเลกุลที่ตื่นเต้น ซึ่งส่งผ่านไปยังสถานะพื้นดินพร้อมกับการปล่อยแสง
เคมีไฟฟ้าประยุกต์
เคมีไฟฟ้ามีการใช้งานจริงมากมาย ด้วยความช่วยเหลือของเซลล์กัลวานิกปฐมภูมิ (องค์ประกอบที่ใช้แล้วทิ้ง) ที่เชื่อมต่อกับแบตเตอรี่ พลังงานเคมีจะถูกแปลงเป็นพลังงานไฟฟ้า แหล่งกำเนิดกระแสทุติยภูมิ - แบตเตอรี่ - เก็บพลังงานไฟฟ้า เซลล์เชื้อเพลิงเป็นแหล่งพลังงานหลักที่ผลิตกระแสไฟฟ้าผ่านการจ่ายสารตั้งต้นอย่างต่อเนื่อง (เช่น ไฮโดรเจนและออกซิเจน) หลักการเหล่านี้รองรับแหล่งพลังงานแบบพกพาและแบตเตอรี่ที่ใช้ในสถานีอวกาศ ยานพาหนะไฟฟ้า และอุปกรณ์อิเล็กทรอนิกส์
การผลิตสารจำนวนมากในปริมาณมากขึ้นอยู่กับการสังเคราะห์เคมีไฟฟ้า อิเล็กโทรไลซิสของน้ำเกลือในกระบวนการคลอร์อัลคาไลจะทำให้เกิดคลอรีนและอัลคาไล ซึ่งจากนั้นจะนำไปใช้ในการผลิตสารประกอบอินทรีย์และโพลีเมอร์ เช่นเดียวกับในอุตสาหกรรมเยื่อกระดาษและกระดาษ ผลิตภัณฑ์จากอิเล็กโทรไลซิสคือสารประกอบ เช่น โซเดียมคลอเรต เปอร์ซัลเฟต โซเดียมเปอร์แมงกาเนต โลหะที่มีความสำคัญทางอุตสาหกรรมได้มาจากการสกัดด้วยไฟฟ้า: อลูมิเนียม แมกนีเซียม ลิเธียม โซเดียม และไทเทเนียม จะดีกว่าถ้าใช้เกลือหลอมเหลวเป็นอิเล็กโทรไลต์เนื่องจากในกรณีนี้ไม่เหมือนกับสารละลายในน้ำตรงที่การลดลงของโลหะนั้นไม่ซับซ้อนโดยการปล่อยไฮโดรเจน ฟลูออรีนผลิตโดยอิเล็กโทรไลซิสในเกลือหลอมเหลว กระบวนการเคมีไฟฟ้าทำหน้าที่เป็นพื้นฐานสำหรับการสังเคราะห์สารประกอบอินทรีย์บางชนิด ตัวอย่างเช่น adiponitrile (ตัวกลางในการสังเคราะห์ไนลอน) ได้มาจากไฮโดรไดเมอไรเซชันของอะคริโลไนไตรล์
การชุบเงิน ทอง โครเมียม ทองเหลือง ทองแดง และโลหะและโลหะผสมอื่นๆ มีการใช้กันอย่างแพร่หลายกับวัตถุต่างๆ เพื่อปกป้องผลิตภัณฑ์เหล็กจากการกัดกร่อน เพื่อวัตถุประสงค์ในการตกแต่ง สำหรับการผลิตขั้วต่อไฟฟ้าและแผงวงจรพิมพ์ในอุตสาหกรรมอิเล็กทรอนิกส์ วิธีเคมีไฟฟ้าใช้สำหรับการประมวลผลขนาดชิ้นงานที่ทำจากโลหะและโลหะผสมที่มีความแม่นยำสูง โดยเฉพาะอย่างยิ่งที่ไม่สามารถแปรรูปด้วยวิธีทางกลแบบเดิมๆ ได้ เช่นเดียวกับการผลิตชิ้นส่วนที่มีโปรไฟล์ที่ซับซ้อน เมื่อพื้นผิวของโลหะ เช่น อลูมิเนียมและไทเทเนียมถูกชุบอโนไดซ์ จะเกิดฟิล์มป้องกันออกไซด์เกิดขึ้น ฟิล์มดังกล่าวถูกสร้างขึ้นบนพื้นผิวของชิ้นงานที่ทำจากอลูมิเนียม แทนทาลัม และไนโอเบียม ในการผลิตตัวเก็บประจุด้วยไฟฟ้า และบางครั้งก็เพื่อการตกแต่ง
วิธีเคมีไฟฟ้ามักใช้ในการศึกษาพื้นฐานของกระบวนการกัดกร่อนและการเลือกวัสดุที่ทำให้กระบวนการเหล่านี้ช้าลง การกัดกร่อนของโครงสร้างโลหะสามารถป้องกันได้โดยใช้การป้องกันแบบแคโทดิก โดยที่แหล่งกำเนิดภายนอกเชื่อมต่อกับโครงสร้างที่ได้รับการป้องกัน และขั้วบวกและโครงสร้างได้รับการบำรุงรักษาให้มีศักยภาพโดยไม่รวมการเกิดออกซิเดชัน กำลังมีการสำรวจความเป็นไปได้ของการประยุกต์ใช้กระบวนการเคมีไฟฟ้าอื่นๆ ในทางปฏิบัติ ดังนั้นอิเล็กโทรไลซิสจึงสามารถนำมาใช้เพื่อทำให้น้ำบริสุทธิ์ได้ ทิศทางที่มีแนวโน้มดีมากคือการแปลงพลังงานแสงอาทิตย์โดยใช้วิธีโฟโตเคมีคอล จอภาพเคมีไฟฟ้ากำลังได้รับการพัฒนา โดยมีหลักการทำงานที่อิงจากเคมีไฟฟ้าเรืองแสง ให้เราพูดถึงการศึกษาการเปลี่ยนแปลงสีของพื้นผิวอิเล็กโทรดแบบพลิกกลับได้อันเป็นผลมาจากปฏิกิริยาอิเล็กโทรด
วรรณกรรม:
อักลาดเซ อาร์.ไอ. เคมีไฟฟ้าประยุกต์ม. – ล., 2518
อิซไมลอฟ เอ็น.เอ. เคมีไฟฟ้าของสารละลายม., 1976
วิธีการวัดในเคมีไฟฟ้าเล่มที่ 1–2. ม., 1977
โคริตา ไอ. ไอออน อิเล็กโทรด เมมเบรนม., 1983
บาโกตสกี้ VS. พื้นฐานของเคมีไฟฟ้าม., 1988