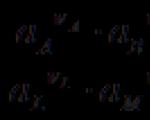Elektrochemie. Galvanische Zelle Elektrochemische Zelle
Elektrochemische Elemente. Elektromotorische Kraft. Thermodynamik einer galvanischen Zelle. EMF-Messung.
Elektrische Doppelschicht, Entstehungsmechanismus und Struktur.
GALVANISCHE ELEMENTE. EMF.
Wenn ein elektrischer Strom durch einen Elektrolyten fließt, kommt es auf der Oberfläche der Elektroden zu elektrochemischen Reaktionen. Das Auftreten elektrochemischer Reaktionen kann durch eine externe Stromquelle erzeugt werden. Auch das umgekehrte Phänomen ist möglich: Elektrochemische Reaktionen, die an zwei in einen Elektrolyten eingetauchten Elektroden ablaufen, erzeugen einen elektrischen Strom, und die Reaktionen laufen nur in einem geschlossenen Stromkreis ab (wenn Strom fließt).
Elektrochemische (oder galvanische) Zelle ist ein Gerät zur Erzeugung von elektrischem Strom durch elektrochemische Reaktionen. Das einfachste elektrochemische Element besteht aus zwei Metallelektroden (Leiter erster Art), die in einen Elektrolyten (Leiter zweiter Art) abgesenkt und durch einen Metallkontakt miteinander verbunden sind. Mehrere in Reihe geschaltete elektrochemische Elemente bilden sich elektrochemischer Kreislauf .
Das wichtigste quantitative Merkmal eines elektrochemischen Elements ist die elektromotorische Kraft(EMF, E), was gleich der Potentialdifferenz ist richtig geöffnetes Element (eines, bei dem Leiter erster Art aus demselben Material mit den Endelektroden des Elements verbunden sind).
Wenn beim Fließen eines elektrischen Stroms in verschiedene Richtungen die gleiche Reaktion auf der Oberfläche der Elektrode auftritt, jedoch in entgegengesetzte Richtungen, werden solche Elektroden sowie das aus ihnen bestehende Element oder der Stromkreis genannt reversibel . Die EMK reversibler Elemente ist ihre thermodynamische Eigenschaft, d.h. hängt nur von T, P, der Art der Substanzen, aus denen die Elektroden und Lösungen bestehen, und der Konzentration dieser Lösungen ab. Ein Beispiel für ein reversibles Element ist Daniel-Jacobi-Element :
(-) Cu çZn çZnSO 4 ççCuSO 4 çCu (+)
bei dem jede Elektrode reversibel ist. Beim Betrieb des Elements treten folgende Reaktionen auf: Zn ® Zn 2+ + 2 e, Cu 2+ + 2 e® Cu. Wenn ein Strom verschwindend geringer Stärke von einer externen Quelle fließt, kommt es zu Rückreaktionen an den Elektroden.
Ein Beispiel für ein irreversibles Element ist Volta-Element :
(-) Zn ç H 2 SO 4 ç Cu (+)
Beim Betrieb des Elements treten folgende Reaktionen auf: Zn ® Zn 2+ + 2 e, 2H + + 2 e® H 2 . Wenn Strom von einer externen Quelle fließt, sind die Elektrodenreaktionen: 2H + + 2 e® H 2 , Cu ® Cu 2+ + 2 e .
Die EMF eines elektrochemischen Elements ist ein positiver Wert, weil es entspricht einem bestimmten spontanen Prozess, der positive Arbeit hervorbringt. Der umgekehrte Vorgang, der nicht selbstständig ablaufen kann, würde einer negativen EMF entsprechen. Beim Aufbau einer Kette elektrochemischer Elemente kann der Prozess in einem der Elemente so gesteuert werden, dass er mit einem Arbeitsaufwand von außen einhergeht (nicht spontaner Prozess), wobei hierfür die Arbeit eines anderen Elements der Kette genutzt wird Dabei kommt es zu einem spontanen Prozess. Die gesamte EMK eines Stromkreises ist gleich der algebraischen Summe positiver und negativer Größen. Daher ist es beim Erstellen eines Schaltplans sehr wichtig, die Vorzeichen der EMF unter Verwendung der anerkannten Regeln zu berücksichtigen.
Die EMK des elektrochemischen Stromkreises gilt als positiv, wenn beim Schreiben der Schaltung die rechte Elektrode relativ zur linken positiv geladen ist (während des Betriebs der Schaltung wandern Kationen in der Lösung von der links geschriebenen Elektrode zur rechts geschriebenen Elektrode und Elektronen wandern hinein die gleiche Richtung im externen Stromkreis). Beispiel.
THERMODYNAMIK EINER GALVANISCHEN ZELLE .
Lassen Sie die Reaktion in einem elektrochemischen System reversibel und isotherm ablaufen:
n A A + n B B + ... ± nF Û n L L + n M M + ... ±
Die vom Element erzeugte elektrische Energie entspricht der Nutzarbeit A¢ des Gesamtprozesses. Die Nutzarbeit A¢ des reversiblen Prozesses ist maximal und entspricht bei P,T = const der Abnahme des isobaren Potentials des Systems:
DG P, T = nFE P, T
E P , T – reversible EMF des Systems.
E P,T = -DG P,T / nF , E V,T = -DF V,T / nF
Durch Messung der EMF des Elements und seines Temperaturkoeffizienten ist es somit möglich, die Werte von DG und DS für den gesamten in der galvanischen Zelle ablaufenden Prozess zu ermitteln. Dieser Prozess ist spontan, daher DG< 0.
Mit der Gibbs-Helmholtz-Gleichung können wir die Enthalpieänderung des Prozesses berechnen:
DH = DG – T = -nFE P + TnF
nFE P = -DH + nFT = + nFT
nFE V = -DU + nFT = + nFT
Aus den Gleichungen folgt, dass der Zusammenhang zwischen der in einem elektrochemischen System reversibel erzeugten oder absorbierten elektrischen Energie und dem thermischen Effekt der darin ablaufenden Reaktion vom Vorzeichen und der Größe des Temperaturkoeffizienten der EMK abhängt dE/ dT :
1. WenndE / dT > 0 , dann ist nFE > (DG > DH) und das System wandelt nicht nur die Wärmemenge, die dem thermischen Effekt der Reaktion entspricht, in elektrische Energie um, sondern auch zusätzliche Wärme - Peletiers Wärme Q P = nFT dE/ dT der Umwelt entlehnt. Unter adiabatischen Bedingungen (unter Bedingungen der Wärmedämmung, wenn ein Austausch mit der Umgebung unmöglich ist) nimmt die T des Systems ab. Die Kühlung des Systems macht sich besonders dann bemerkbar, wenn dE/ dT > 0 < 0 (реакция эндотермична).
2. WenndE / dT < 0 , dann nFE< (DG < DH) и часть теплоты реакции будет рассеиваться в виде теплоты Пелетье. В адиабатическом режиме система будет нагреваться.
3. WenndE / dT = 0 , dann ist DG = DH und nFE = – die vom System reversibel erzeugte elektrische Energie entspricht dem thermischen Effekt der chemischen Reaktion. Diese Beziehung ist bekannt als Thomsons Prinzip (Regel) .
Um die EMF zu berechnen, können die Gleichungen wie folgt umgeschrieben werden:
Bei der Verwendung von Gleichungen muss beachtet werden, dass sie gilt nur für reversible elektrochemische Systeme Daher ist es bei der Untersuchung der EMF-Abhängigkeit von T notwendig, die Verwendung elektrochemischer Systeme mit Flüssigkeitsgrenzen zu vermeiden, weil die auf ihnen entstehenden Diffusionspotentiale sind nicht im Gleichgewicht.
Verknüpfen wir die EMF des Elements mit der Gleichgewichtskonstante der im Element ablaufenden Reaktion. Isothermengleichung einer chemischen Reaktion:
DG = RT ln K A -RT
E = - = ln K A -
Der erste Term auf der rechten Seite der Gleichung ist für gegebenes P, T ein konstanter Wert; er kann mit E o bezeichnet werden. E o - Standard-EMF eines Elements (elektrochemisches System) , d.h. EMF überhaupt ein i = 0.
E = E o + ln
 = E o + 2,303 lg
= E o + 2,303 lg
Somit ist die EMF eines elektrochemischen Systems eine Funktion der Aktivitäten der Teilnehmer an der elektrochemischen Reaktion. Die obigen Gleichungen ermöglichen die Berechnung der Werte von DG und K A basierend auf experimentellen Werten von E und berechnen Sie umgekehrt E, wobei Sie die thermodynamischen Eigenschaften der chemischen Reaktion kennen.
EMF-MESSUNG .
Um den Gleichgewichtswert (reversiblen Wert) der EMF eines elektrochemischen Elements zu messen, ist es notwendig, dass der Prozess unendlich langsam abläuft, d.h. so dass das Element mit einem verschwindend kleinen Strom arbeitet. Diese Bedingung wird bei der Kompensationsmethode erfüllt, die darauf basiert, dass das Element in Reihe gegen eine äußere Potentialdifferenz geschaltet wird und diese so gewählt wird, dass kein Strom im Stromkreis fließt. Dann ist die externe Potentialdifferenz gleich der EMK des Stromkreises.
Mit der Kompensationsmethode können Sie den Wert der EMF direkt messen. Dies ist jedoch ein ziemlich komplizierter Vorgang. Daher wird in der Laborpraxis lieber die EMF des untersuchten Elements mit der EMF des sogenannten Standards (normal) verglichen. Elemente, die sorgfältig bei verschiedenen T gemessen werden. Diese Vergleichsmethode ist auch eine Kompensation.
Das grundlegende normale Element ist gesättigtes Weston-Element .
(EMF-Messkreis – unabhängig).
STRUKTUR DER ELEKTRODENGRENZE - LÖSUNG. DOPPELTE ELEKTRISCHE SCHICHT .
Wenn ein Leiter erster Art mit einem Elektrolyten in Kontakt kommt, a elektrische Doppelschicht . Betrachten Sie als Beispiel eine Kupferelektrode, die in eine CuSO 4 -Lösung eingetaucht ist. Das chemische Potenzial von Kupferionen in einem Metall bei einer gegebenen T kann als konstant angesehen werden, während das chemische Potenzial von Kupferionen in Lösung von der Salzkonzentration abhängt; Im Allgemeinen sind diese chemischen Potenziale nicht gleich.
Die Konzentration von CuSO 4 sei so, dass > . Wenn das Metall dann in die Lösung eingetaucht wird, werden einige der Cu 2+-Ionen aus der Lösung dehydriert und auf das Metall übertragen, wodurch eine positive Ladung darauf entsteht. Diese Ladung verhindert die weitere Übertragung von Cu 2+-Ionen aus der Lösung auf das Metall und führt zur Bildung einer Schicht aus SO 4 2--Anionen, die in der Nähe der Elektrode angezogen werden. Die sogenannte elektrochemisches Gleichgewicht , bei dem sich die chemischen Potentiale der Ionen im Metall und in der Lösung um die Größe der Potentialdifferenz der resultierenden doppelten elektrischen Schicht (DEL) unterscheiden:
Im elektrochemischen Gleichgewicht gleichen sich die elektrische Potentialdifferenz und die chemische Potentialdifferenz aus.
Lassen Sie die Konzentration von CuSO 4 so niedrig sein, dass< . В этом случае при погружении металла в раствор будет наблюдаться обратный процесс перехода ионов меди из кристаллической решетки металла в раствор и электрод окажется заряженным отрицательно. Этот заряд будет препятствовать дальнейшему переходу ионов Cu 2+ в раствор, установится новое электрохимическое равновесие.
Sie können eine Elektrolytkonzentration wählen, bei der die chemischen Potentiale der Ionen im Metall und in der Lösung gleich sind. Lösungen dieser Konzentration heißen Null Lösungen . Wenn ein Metall in seine Nulllösung eingetaucht wird, tritt kein EDL auf der Oberfläche der Elektrode auf; allerdings ist auch in diesem Fall die Potentialdifferenz zwischen dem Metall und der Lösung nicht Null.
Laut Nernst ist die einzige EMF-Quelle einer elektrochemischen Zelle die EMF auf der Oberfläche der Elektroden. Nernst definierte das Potential von Metallen in einer Nulllösung als absolutes Nullpotential. In den Werken von A. N. Frumkin wurde gezeigt, dass Nernsts Ideen falsch sind. Es wurde experimentell festgestellt, dass die EMF eines Elements, das aus zwei verschiedenen Elektroden besteht, die in ihre Nulllösungen eingetaucht sind, sehr deutlich von Null abweicht (vielleicht mehr als 1 V). Das Potential eines Metalls in einer Nulllösung, genannt Nullladungspotential , kann nicht als absolutes Nullpotential betrachtet werden.
THEORIE DER KONDENSIERTEN HELMHOLTZ-DOPPELSCHICHT. Die erste quantitative Theorie der Struktur von DEL an der Grenzfläche Metall-Lösung wurde von Helmholtz (1853) erstellt. Laut Helmholtz kann ein EDL mit einem flachen Kondensator verglichen werden, dessen eine Platte mit der Ebene durch die Oberflächenladungen im Metall zusammenfällt, die andere mit der Ebene, die die Ladungszentren der Ionen in der Lösung verbindet. Durch elektrostatische Kräfte werden sie von der Metalloberfläche angezogen. Doppelte Schichtdicke l gleich dem Ionenradius R. Gemäß der Bedingung der elektrischen Neutralität muss die Anzahl der von der Metalloberfläche angezogenen Ionen so groß sein, dass ihre Ladungen die Oberflächenladungen des Metalls kompensieren, d. h.
Die Theorie einer kondensierten Doppelschicht ermöglicht es, experimentell konsistente Werte der EDL-Kapazität und eine physikalisch plausible Dicke der EDL zu erhalten. Es kann jedoch viele experimentelle Gesetzmäßigkeiten nicht interpretieren: die experimentell gefundenen Werte des elektrokinetischen Potentials (x-Potenzial) und deren Abhängigkeit von der Elektrolytkonzentration, die Änderung des Vorzeichens der Ladung auf der Metalloberfläche in Gegenwart eines Tensids .
THEORIE DER DIFFUSEN DOPPELSCHICHTEN-GUI- CHAPMAN. Die Theorie von Helmholtz berücksichtigt nicht, dass sich die Eigenschaften des DES mit der Konzentration des Elektrolyten ändern, und T. Gouy (1910) und Chapman (1913) versuchten, die Ladungsdichte im DES mit der Zusammensetzung der Lösung in Beziehung zu setzen. Sie berücksichtigten, dass neben den elektrostatischen Kräften, die zwischen dem Metall und den Ionen entstehen, auch die Kräfte der thermischen Molekülbewegung auf die Ionen einwirken. Wenn diese beiden Kräfte wirken, sollten die Ionen in der Lösung relativ zur Metalloberfläche diffus verteilt sein – wobei die volumetrische Ladungsdichte mit der Entfernung von ihr abnimmt.
Gouy und Chapman glaubten, dass Ionen als materielle Punkte betrachtet werden können, die kein eigenes Volumen, aber eine Ladung haben, und dass ihre Verteilung im Elektrodenladungsfeld der Boltzmann-Verteilung gehorcht.
Die Gouy-Chapman-Theorie stimmt besser mit den Gesetzen elektrokinetischer Phänomene überein als die Helmholtz-Theorie. Gehen wir davon aus, dass ab einer bestimmten Entfernung l 1 Ionen bei relativer Bewegung der festen und flüssigen Phase nicht mehr fest an die Elektrodenoberfläche gebunden sind, dann kann das diesem Abstand entsprechende Potential als x-Potential (x) betrachtet werden< j). Однако теория не объясняет изменение знака x-потенциала и перезарядку поверхности с изменением состава раствора. Кроме того, теория Гуи-Чапмана оказывается менее удовлетворительной, чем теория Гельмгольца, при использовании ее для количественных расчетов емкости ДЭС, т.к. она не учитывает собственного объема ионов, которые отождествляются с материальными точками.
Somit lässt sich die Gouy-Chapman-Theorie am besten dort rechtfertigen, wo sich die Helmholtz-Theorie als nicht anwendbar erweist, und umgekehrt liefert letztere eine bessere Konvergenz mit dem Experiment in Fällen, in denen erstere falsche Ergebnisse liefert. Folglich muss die Struktur des DES einer Kombination der von Helmholtz und Gouy-Chapman vorgeschlagenen Modelle entsprechen. Diese Annahme wurde von Stern (1924) in seiner Adsorptionstheorie von DEL gemacht.
STERN-ADSORPTIONSTHEORIE. Stern glaubte, dass ein bestimmter Teil der Ionen in der Nähe der Metall-Elektrolyt-Grenzfläche zurückgehalten wird und eine Helmholtz- oder kondensierte Doppelschichtplatte bildet, deren Dicke dem durchschnittlichen Radius der Elektrolytionen entspricht. Die restlichen im EDL enthaltenen Ionen verteilen sich diffus mit allmählich abnehmender Ladungsdichte. Für den diffusen Teil des EDL vernachlässigte Stern wie Gouy die intrinsische Größe der Ionen. Darüber hinaus schlug Stern vor, dass im dichten Teil der EDL Ionen nicht nur aufgrund elektrostatischer Kräfte, sondern auch spezifischer Adsorptionskräfte, d. h. Kräfte nicht-coulombschen Ursprungs. Daher kann in Lösungen, die oberflächenaktive Ionen enthalten, deren Anzahl im dichten Teil der EDL die Ladung der Metalloberfläche um einen bestimmten Betrag übersteigen, abhängig von den Eigenschaften der Ionen und der Ladung des Metalls. Somit sind laut Stern zwei Modelle von DES zu unterscheiden, von denen sich das eine auf Lösungen oberflächeninaktiver Elektrolyte bezieht, das andere auf Lösungen, die spezifisch adsorbierte Ionen enthalten.
Auch in der Adsorptionstheorie bleibt die Gleichheit erhalten:
Q M = q L = q 1 + q 2
Die Ladungsdichte auf der Lösungsseite q L besteht aus zwei Teilen: der Ladungsdichte in der Helmholtz-Schicht q 1 und der Ladungsdichte in der diffusen Schicht q 2 .
Sterns Theorie ermöglicht es uns, das x-Potenzial als den Potentialabfall im diffusen Teil der EDL zu definieren, wo die starke Bindung zwischen dem Metall und den Ionen bereits verloren gegangen ist. Bei dieser Definition sollte das x-Potenzial nicht mit dem Nerst-Potential zusammenfallen, wie experimentell beobachtet wird. Sterns Theorie konnte die Neuaufladung der Oberfläche eines Festkörpers erklären.
Bei einer verschwindend kleinen Konzentration sind alle Ladungen in der Lösung diffus verteilt und die Struktur der EDL wird durch die Gouy-Chapman-Theorie beschrieben. Im Gegenteil: In konzentrierten Lösungen nähert sich die Struktur von DES dem von Helmholtz vorgeschlagenen Modell an. Im Bereich der durchschnittlichen Konzentrationen, in denen x in seiner Größe mit RT/F vergleichbar ist, kann seine Abhängigkeit von der Konzentration durch Näherungsgleichungen ausgedrückt werden:
für positive Werte x: x = B - ln Mit
für negative Werte von x: x = B¢ + ln Mit
Sterns Theorie liefert ein qualitativ korrektes Bild der DEL. Die Bestimmung der Kapazität mit dem Stern-Modell entspricht sowohl hinsichtlich der Kapazitätswerte als auch hinsichtlich der Art ihrer Abhängigkeit vom Elektrodenpotential und der Lösungskonzentration der Erfahrung. Doch Sterns Theorie ist nicht frei von Mängeln. Dazu gehört die Unmöglichkeit einer quantitativen Beschreibung von Kapazitätsverläufen, insbesondere bei der Abkehr vom Nullladungspotential.
WEITERENTWICKLUNG DER THEORIE DES STANDINGS. Es wurden viele Versuche unternommen, eine DES-Theorie zu entwickeln, die quantitativ mit experimentellen Daten übereinstimmt (Rice, Frumkin et al., Bockris, Devanathan, Esin, Muller, Parsons, Ershler usw.). Das am weitesten verbreitete Modell ist Graham (1947). Laut Graham besteht die DES-Beschichtung in der Lösung nicht aus zwei, sondern aus drei Teilen. Die erste, von der Oberfläche des Metalls aus gezählt, wird als innere Helmholtz-Ebene bezeichnet; es enthält nur oberflächenaktive Ionen (die Ladung der Ebene ist q 1) oder, wenn sie nicht in der Lösung sind, Lösungsmittelmoleküle (q 1 = 0); sein auf die Lösung bezogenes Potential wird mit y 1 bezeichnet. Die nächste, von der Metalloberfläche in einer Entfernung entfernt, bis zu der sich Ionen (ihre Ladungszentren) nähern können, wird äußere Helmholtz-Ebene genannt; seine Gesamtladung ist q 2 und das Potential der Ebene ist y 2. Hinter der äußeren Helmholtz-Ebene befindet sich eine diffuse Schicht mit einem Potential zwischen y 2 und Null und einer Ladungsdichte, die mit q 2 übereinstimmt.
Grahams Modell spiegelt die Hauptmerkmale und Eigenschaften der Metall-Elektrolyt-DES-Struktur wider. Damit können Sie Differentialkapazitätskurven für jede Konzentration eines bestimmten Elektrolyten berechnen, wenn für mindestens eine seiner Lösungen eine experimentelle Kurve vorliegt. Allerdings deckt dieses Modell nicht alle Aspekte des Problems ab.
Wenn ein elektrischer Strom durch eine Lösung fließt, fließen Ströme auf der Oberfläche der Elektroden. Elektrochemische Reaktionen, die mit dem Fluss von Elektronen zur oder von der Elektrode einhergehen. Bei umgekehrten Prozessen führen elektrochemische Reaktionen an den Grenzflächen zwischen Leitern erster und zweiter Art zur Erzeugung eines elektrischen Stroms.
Elektrochemische Prozesse unterscheiden sich in einigen Merkmalen von herkömmlichen chemischen Reaktionen.
Eine chemische Reaktion ist nur möglich, wenn reagierende Teilchen kollidieren. Wenn sie in Kontakt kommen, können Elektronen von einem Teilchen auf ein anderes übertragen werden. Ob ein solcher Übergang tatsächlich stattfindet, hängt von der Energie der Teilchen und ihrer gegenseitigen Ausrichtung ab. Die Aktivierungsenergie hängt von der Art der chemischen Reaktion ab und ist bei ionischen Reaktionen normalerweise niedrig. Der Elektronenübergangsweg ist sehr kurz, was auch ein Merkmal der chemischen Reaktion ist. Kollisionen von Teilchen können an beliebigen Punkten des Reaktionsraums an unterschiedlichen gegenseitigen Positionen auftreten, daher können elektronische Übergänge in beliebige Richtungen erfolgen, d. h. Merkmale des chemischen Prozesses sind die Zufälligkeit von Kollisionen und die fehlende Richtungsabhängigkeit elektronischer Übergänge. Dadurch treten die energetischen Wirkungen chemischer Reaktionen vor allem in Form von Wärme in Erscheinung (eine geringe Ausdehnungsarbeit ist ebenfalls möglich).
Damit sich die einer chemischen Umwandlung entsprechenden Energieänderungen in Form von elektrischer Energie, d. h. Damit der elektrochemische Prozess ablaufen kann, ist es notwendig, die Reaktionsbedingungen zu ändern.
Elektrische Energie ist immer mit dem Durchgang von elektrischem Strom verbunden, d.h. Elektronenfluss in eine bestimmte Richtung. Daher muss die Reaktion so durchgeführt werden, dass die elektronischen Übergänge nicht zufällig sind, sondern in eine Richtung erfolgen, und ihr Weg muss deutlich größer als die Atomgröße sein. Daher muss bei elektrochemischen Prozessen der Übergang von Elektronen von einem Teilnehmer zum anderen in beträchtlicher Entfernung erfolgen, wofür eine räumliche Trennung der Reaktionsteilnehmer erforderlich ist. Eine räumliche Trennung allein reicht jedoch nicht aus, da die Reaktion dadurch einfach zum Stillstand kommt.
Um den elektrochemischen Prozess durchzuführen, sind zusätzliche Bedingungen notwendig: Elektronen müssen von einigen Teilchen abgerissen und auf eine gemeinsame Weise auf andere übertragen werden. Dies kann erreicht werden, indem der direkte Kontakt zwischen den Reaktionsteilnehmern durch ihren Kontakt mit zwei Metallen ersetzt wird, die durch einen Metallleiter miteinander verbunden sind. Damit der Elektronenfluss kontinuierlich ist, ist es auch notwendig, den Durchgang von elektrischem Strom durch den Reaktionsraum sicherzustellen, der in der Regel von den Teilnehmern der elektrochemischen Reaktion selbst (sofern sie sich in einem ionisierten Zustand befinden) oder durchgeführt wird durch spezielle Verbindungen mit hoher Ionenleitfähigkeit.
Man bezeichnet ein Gerät zur Erzeugung elektrischer Energie durch elektrochemische Reaktionen elektrochemisch(oder galvanisch)Element. Das einfachste elektrochemische Element besteht aus zwei Metallelektroden (Leiter erster Art), die in eine Elektrolytlösung (Leiter zweiter Art) getaucht sind.
Wenn beim Fluss eines elektrischen Stroms in verschiedene Richtungen die gleiche Reaktion auf der Oberfläche der Elektrode auftritt, jedoch in entgegengesetzte Richtungen, werden solche Elektroden sowie daraus zusammengesetzte elektrochemische Elemente genannt reversibel. Ein Beispiel für ein reversibles Element ist das Daniel-Jacobi-Element
(–) Zn | ZnSO 4, Lösung || CuSO 4, Lösung | Cu(+)
Beim Betrieb eines solchen Elements kommt es an den Elektroden zu elektrochemischen Reaktionen:
Zn Zn 2 + + 2e
Cu 2 + + 2eCu
Die Gesamtreaktionsgleichung in einem Element kann dargestellt werden als:
Zn + Cu 2 + Zn 2 + + Cu
Wenn ein verschwindend kleiner Strom von einer externen Quelle durch das Element fließt, laufen diese Reaktionen in die entgegengesetzte Richtung ab.
Beispiel irreversibel Element ist das Volta-Element
(–) Zn | H2SO4 | Cu(+)
Beim Betrieb eines solchen Elements treten an den Elektroden folgende Reaktionen auf:
Zn Zn 2 + + 2e
2H + + 2eH 2 ,
und die Reaktion im Element wird durch die Gleichung dargestellt
Zn + 2H + Zn 2+ + H 2
Wenn Strom von einer externen Quelle fließt, treten an den Elektroden andere Reaktionen auf:
Cu Cu 2 + + 2e,
diese. In einer elektrochemischen Zelle löst sich Kupfer in Schwefelsäure unter Freisetzung von Wasserstoff:
Cu + 2H + Cu 2 + + H 2
Das wichtigste Merkmal einer elektrochemischen Zelle ist ihre elektromotorische Kraft(EMF) E– Potentialdifferenz eines richtig geöffneten Elements, d.h. die Potentialdifferenz zwischen den Enden von Leitern erster Art aus demselben Material, die mit den Elektroden einer galvanischen Zelle verbunden sind. Mit anderen Worten, EMF ist die Potentialdifferenz unter Gleichgewichtsbedingungen, wenn im Stromkreis kein elektrischer Strom fließt. Wenn Sie die Elektroden schließen, fließt ein elektrischer Strom im Stromkreis und stellt die Potentialdifferenz dar Stromspannung ein elektrochemisches Element, das sich von der EMF durch den Spannungsabfall am Innenwiderstand des Elements unterscheidet.
Der Zweck elektrochemischer Elemente besteht darin, die Bewegung von Elektronen im externen Stromkreis zu fördern, in dem das nützliche Element enthalten ist
Belastung. Um ihre Aufgabe zu erfüllen, muss eine Brennstoffzelle daher eine Elektronenquelle und eine Elektronensenke enthalten (Abb. 7.1).
Die in einer elektrochemischen Zelle ablaufenden Reaktionen werden Reduktionsreaktionen genannt, da der Begriff „Oxidation“ dem Prozess der Freisetzung von Elektronen und der Begriff „Reduktion“ dem Prozess der Freisetzung von Elektronen entspricht.
Die Bedeutung zahlreicher veralteter wissenschaftlicher Konzepte lässt sich oft nicht aus ihren Namen ersehen. Die Begriffe „Oxidation“ und „Reduktion“ bedürfen einer Erklärung. Im Südosten kommt Sauerstoff (Sauerstoff) von der lateinischen Wurzel oxus (sauer oder scharf, scharf) und bedeutet „Säureerzeuger“. Dieser Name wurde erstmals in der 1787 veröffentlichten Arbeit „Nomenclature Chimique“ von Morveau und Lavoisier verwendet. Damals hielten Chemiker an einem Irrglauben fest. dass Sauerstoff das Hauptelement von Säuren ist; wenn eine Säure in Wasser gelöst wird, verliert ein Teil der in ihrer Zusammensetzung enthaltenen Wasserstoffatome ihr Elektron – das Wasser wird sauer und der Wasserstoff wird oxidiert. Analytisch gesehen wird jede Reaktion, die mit dem Prozess des Elektronenverlusts verbunden ist, als Oxidation bezeichnet. Die Rückreaktion – die Elektroneneinfangreaktion – wird „Werden“ genannt.
Fließen
J-Elektronen
Elektronenquelle >
(Oxidation)
Positiv< -
Richtung des elektrischen Stroms Abb. 7.1. Eine elektrochemische Zelle muss eine Elektronenquelle und eine Elektronensenke enthalten
In einer elektrochemischen Zelle wird die Gesamtreaktion in zwei Zwischenreaktionen aufgeteilt, die in getrennten Bereichen des Geräts ablaufen. Diese Bereiche werden durch den Elektrolyten verschmolzen, der ein Ionenleiter ist, aber die bei der Zwischenoxidationsreaktion freigesetzten Elektronen leitet. Elektronen können in den Bereich gelangen, in dem die Reduktionsreaktion stattfindet, jedoch über einen externen Stromkreis. Somit entsteht im externen Stromkreis ein elektrischer Strom, und die elektrochemische Zelle ist seine Quelle – das ist ihr Zweck. Als positive Richtung des elektrischen Stroms a im externen Stromkreis wurde die Richtung vom reduzierenden Oxidationsbereich des Elements vereinbart – der elektrische Strom verlässt das Element des Reduktionsbereichs, der somit die Kathode des Brennstoffs ist – zum Element und gelangt in den Oxidationsbereich, der die Anode darstellt. Wie bei jeder anderen Stromquelle ist die Kathode positiv geladen
aktive Elektrode und die Anode ist eine negativ geladene Elektrode. Bei Verbrauchern elektrischer Energie (Last) ändern sich die Bezeichnungen ins Gegenteil. In der Einleitung zu Kap. 6 Auf die Begriffe „Anode“ und „Kathode“ wird näher eingegangen.
Betrachten Sie als Beispiel einer elektrochemischen Zelle eine Membran, die die Rolle eines Elektrolyten spielt. Lassen Sie eine der Seitenflächen der Membranen mit Wasserstoff in Kontakt stehen. Unter normalen Bedingungen besteht das Gas überwiegend aus molekularem Wasserstoff, eine kleine Anzahl von Molekülen kann jedoch in Atome dissoziieren
und einige der Atome werden oxidieren (ionisieren), d. h. ihr Elektron verlieren
N -> Nt + e~ .
Da die Membran für Elektronen undurchlässig ist, bleiben sie alle auf einer Seite davon, während die gebildeten Ionen durch Diff; Durch die Membran gelangen sie auf die andere Seite. In diesem Fall tragen die Ionen eine positive Ladung, sodass die Oberfläche der Membran, auf der sich Wasserstoff befindet, aufgrund des Überschusses an darauf angesammelten Elektronen negativ geladen wird und die gegenüberliegende Oberfläche aufgrund des Positiven positiv geladen wird Ionen, die durch Diffusion darauf entstanden sind. Das elektrische Feld bewirkt, dass sich einige der Ionen in die entgegengesetzte Richtung zur „Wasserstoff“-Oberfläche der Membran bewegen. Das dynamische Gleichgewicht im betrachteten System stellt sich ein, wenn der Diffusionsfluss der Ionen st. i ist gleich dem Rückstrom.
Tragen wir nun elektrisch leitfähiges Pulver auf beide Oberflächen der Membran auf. Dadurch entstehen zwei poröse elektrisch leitfähige Schichten, die als Elektroden fungieren. Schließen wir eine externe Heizung1 an die Elektroden an und stellen so die elektrische Verbindung der Elektroden sicher. Belastungswiderstand bis erheblichem Rl. Ionen können sich im externen Kreislauf nicht bewegen, und Elektronen beginnen, sich vom „Wasserstoff“-Teil, wo ein Überschuss vorhanden ist, auf die gegenüberliegende Seite der Membran zu bewegen und bilden im externen Kreislauf einen elektrischen Strom, wie in Abb. 7.2. Die für uns interessante Reaktion findet an der Elektrode „Wasser“ statt und wird durch die Gleichung beschrieben
2H2 -> 4H+ + 4e (anodische Reaktion)
Das beschriebene Funktionsschema des Elements hat einen wesentlichen Nachteil: Es widerspricht dem ersten Hauptsatz der Thermodynamik. Wenn tatsächlich ein elektrischer Strom durch eine externe Last fließt, wird die Menge davon freigesetzt, deren Menge durch das Produkt I2RL bestimmt wird. Gleichzeitig elektrisch
Beim Eintritt in den Kathodenbereich verbinden sie sich mit H+-Ionen, die durch die Membran diffundieren, Wasserstoffatome bilden und schließlich H2-Gasmoleküle erzeugen, die als „Brennstoff“ verwendet werden. Würde der Prozess nach dem vorgegebenen Schema ablaufen, würden wir Wärme ohne Brennstoffverbrauch erhalten.
Der äußere Stromkreis bildet nur einen Weg für die Bewegung von Elektronen, kann aber an sich nicht die Ursache für die Entstehung von elektrischem Strom sein. Genauso wie wenn man ein Ende eines Rohrs in einen See senkt, fließt das Wasser selbst nicht in das Rohr. Damit Wasser fließen kann, muss sich das andere Ende unter der Wasseroberfläche befinden. Ebenso ist es zum Einleiten eines elektrischen Stroms in einen externen Stromkreis erforderlich, das rhmodynamische Potenzial im Kathodenbereich zu verringern. Am einfachsten gelingt dies durch Zugabe von Sauerstoff, dessen Moleküle sich mit Elektronen und Ionen verbinden und so Wasser entsteht:
4е“ + 4Н+ + 02 -» 2Н20 (Kathodenreaktion). (4)
Die Reaktion ist exotherm, d. h. sie verläuft unter Freisetzung überwiegend elektrischer Energie und nicht thermischer Energie, wie dies bei der Verbrennung von Wasserstoff der Fall ist. Offensichtlich ist es diese Energie, die das Brennelement antreibt.
Das beschriebene Diagramm der elektrochemischen Zelle ist in Abb. dargestellt. 7.2.
Unter normalen Bedingungen ist der Anteil der dissipierten Wasserstoffmoleküle* im gesamten Bereich gering. Sie kann durch Änderung der physikalischen Parameter leicht erhöht werden
Meter nach dem Prinzip von Le Chatelier. zum Beispiel durch Erhöhung der Systemtemperatur. Der Dissoziationsgrad von Wasserstoffmolekülen kann auch durch die Wirkung von Katalysatoren erhöht1 werden.
Die vollständige Reaktion, die in einer elektrochemischen Zelle abläuft, wird durch die Gleichung beschrieben:
2H2 + 02 -> 2H?0.
Die elektrochemische Zelle, erfunden von Alessandro Volta (1745–1800), war das erste Gerät zur kontinuierlichen Erzeugung von elektrischem Strom. Sie bestand aus Zink- und Silber- (oder Kupfer-) Basen, die durch in einer Salzlösung getränkte Papierblätter getrennt waren Die galvanische Batterie entstand durch Reihenschaltung der Elemente, wobei die Elektrode eines Elements direkt mit der ZINK-Elektrode eines anderen Elements verbunden wurde.
Eine elektrochemische Volta-Zelle kann durch Eintauchen von Schweiß- und Kupferelektroden in eine verdünnte (z. B. 10 %) Lösung dieser Säure hergestellt werden. Zink oxidiert:
Zn -> Zr,++ + 2e >
Dadurch entstehen freie Elektronen. Zinkionen in Wasser auflösen. Schwefelsäure ist eine starke Säure1) und zerfällt in Ionen:
H2S04 - „2Н+ + SOf“
Zinkionen verbinden sich mit Sulfationen zu Zinksulfat. Pons in Form des Hydroniumions H" (H20)x bewegen sich durch den Elektrolyten zur Elektrode, an der sie in Form von Gasblasen freigesetzt werden (wenn das Hydroniumion Elektronen einfängt, die von außerhalb des II zur Elektrode gelangen). Dieser Typ einer elektrochemischen Zelle ist in der Praxis nahezu unbrauchbar, da die Kupferelektrode sehr schnell mit Wasserstoffblasen bedeckt wird, die an ihrer Oberfläche haften, was den Fluss der in die Elektrode eindringenden Ionen erheblich verringert. Sogenannte „Trockenzellen“ verwenden eine Methode, um die Bildung einer isolierenden Gasschicht auf der Oberfläche zu verhindern. Das chemische Reagenz, das zur Absorption des Wasserstoffs verwendet wird, wird Depolarisator genannt. Eines der verbrauchten Reagenzien ist normalerweise
"> Die Stärke einer Säure wird durch den Dissoziationsgrad ihrer Moleküle in einer wässrigen Lösung bestimmt. C-Säure dissoziiert vollständig in H+- und C1_-Ionen – das ist eine starke Säure. Schwefelsäure ist etwas schwächer, gehört aber ebenfalls zu den starken Säuren Überraschenderweise ist Flusssäure trotz ihrer hohen Korrosivitätsaktivität eine schwach saure Lösung – bei Raumtemperatur beträgt der Anteil der H+-Ionen an der Gesamtkonzentration der Neutra-HF-Moleküle weniger als 3 %.
Galle, die leicht oxidiert; Am häufigsten wird Zink verwendet. Beachten Sie, dass die Kupferelektrode im Volta-Element keine chemischen Reaktionen eingeht und daher kein Kupfer verbraucht wird.
Bis vor Kurzem wurden in kostengünstigen galvanischen Batterien Leclanchet-Zellen verwendet, bei denen die Anode aus Zink und die Kathode aus einem Graphitstab besteht, der mit einer Schicht aus pulverisiertem Mangandioxid unter Zusatz von Graphit (zur Erhöhung der elektrischen Leitfähigkeit) beschichtet ist. Mangandioxid absorbiert den freigesetzten Wasserstoff und verhindert so die Bildung einer Gasschicht auf der Kathodenoberfläche. Als Elektrolyt wird Ammoniumchlorid verwendet. Moderne (Alkali-)Batterien verwenden einen alkalischen Elektrolyten.
Wäre das für das Anodenmaterial verwendete Zink absolut rein, würde es nur verbraucht, wenn elektrischer Strom durch das Element fließt. Das Vorhandensein von Verunreinigungen führt auch dann zur Korrosion der Elektrode, wenn das Element nicht verwendet wird (Verunreinigungen bilden zahlreiche mikroskopisch kleine elektrochemische Zellen im Inneren des Elektrodenmaterials). Um eine lange Haltbarkeit solcher Batterien zu gewährleisten, besteht die Anode aus einer Legierung aus Zink und Quecksilber (amalgamiert). Derzeit wurden Leclanche-Zellen fast vollständig durch Alkalibatterien ersetzt.
Viele chemische Reaktionen laufen nur ab, wenn Energie von außen zugeführt wird. Sie werden häufig in Elektrolysezellen (Elektrolyseuren) an Elektroden durchgeführt, die an eine externe Stromquelle angeschlossen sind. Die Untersuchung dieser Reaktionen liefert Informationen über die Natur und Eigenschaften verschiedener Stoffe und ermöglicht auch die Gewinnung neuer chemischer Verbindungen durch Elektrosynthese. Elektrochemische Verfahren sind in der Industrie weit verbreitet. Beispiele hierfür sind die Chlor- und Aluminiumproduktion, die Galvanisierung und die elektrische Gewinnung. Galvanische Zellen, die chemische Energie in elektrische Energie umwandeln, bilden die Grundlage für Stromquellen – Batterien und Akkumulatoren sowie Brennstoffzellen. Die Elektrochemie untersucht auch andere elektrische Phänomene: das Verhalten von Ionen in Elektrolytlösungen und den Stromdurchgang durch solche Lösungen; Trennung von Ionen in einem elektrischen Feld (Elektrophorese); Korrosion und Passivierung von Metallen; elektrische Effekte in biologischen Systemen (Bioelektrochemie); photoelektrochemische Prozesse (die Wirkung von Licht auf elektrochemische Reaktionen in Zellen).
Historische Referenz.
Erst nach der Schaffung einer konstanten, ausreichend leistungsstarken elektrischen Stromquelle wurde es möglich, systematische elektrochemische Forschungen durchzuführen. Eine solche Quelle tauchte an der Wende vom 18. zum 19. Jahrhundert auf. als Ergebnis der Arbeit von L. Galvani und A. Volta. Während Galvani die physiologischen Funktionen des Frosches untersuchte, schuf er versehentlich einen elektrochemischen Schaltkreis, der aus zwei verschiedenen Metallen und dem Muskel eines sezierten Froschschenkels bestand. Wenn die Pfote, die mit einem Kupferhalter befestigt war, mit einem Eisendraht berührt wurde, der ebenfalls mit dem Halter verbunden war, zog sich der Muskel zusammen. Ähnliche Kontraktionen traten unter dem Einfluss einer elektrischen Entladung auf. Galvani erklärte dieses Phänomen mit der Existenz „tierischer Elektrizität“. Eine andere Interpretation dieser Experimente gab Volta, der glaubte, dass Elektrizität am Kontaktpunkt zweier Metalle entsteht und die Kontraktion des Froschmuskels das Ergebnis des Durchgangs von elektrischem Strom durch ihn ist. Ein Strom entstand auch, wenn ein mit Salzwasser getränktes schwammiges Material (Tuch oder Papier) zwischen zwei Metallscheiben, zum Beispiel Zink und Kupfer, gelegt und der Stromkreis geschlossen wurde. Durch die Reihenschaltung von 15–20 dieser „Elemente“ schuf Volta im Jahr 1800 die erste chemische Stromquelle – die „Voltaische Säule“.
Der Einfluss von Elektrizität auf chemische Systeme interessierte sofort viele Wissenschaftler. Bereits im Jahr 1800 berichteten W. Nicholson und A. Carlyle, dass Wasser in Wasserstoff und Sauerstoff zerfällt, wenn ein elektrischer Strom mithilfe von Platin- und Golddrähten, die an eine „Voltasäule“ angeschlossen sind, durch das Wasser geleitet wird. Die wichtigste der frühen elektrochemischen Studien war die Arbeit des englischen Chemikers H. Davy. 1807 isolierte er das Element Kalium, indem er einen Strom durch leicht angefeuchtetes festes Kaliumhydroxid leitete. Als Spannungsquelle diente eine Batterie aus 100 galvanischen Zellen. Auf ähnliche Weise wurde metallisches Natrium gewonnen. Davy isolierte später Magnesium, Kalzium, Strontium und Barium durch Elektrolyse mit einer Quecksilberelektrode.
Davys Assistent M. Faraday untersuchte den Zusammenhang zwischen der Strommenge (Strom mal Zeit), die durch die Grenzfläche zwischen Elektrode und Lösung fließt, und den dadurch verursachten chemischen Veränderungen. Ein Instrument (heute bekannt als Gascoulometer) wurde entwickelt, um die Elektrizitätsmenge aus dem in einer Elektrolysezelle freigesetzten Wasserstoff- und Sauerstoffvolumen zu messen, und es wurde gezeigt (1833), dass die Elektrizitätsmenge erforderlich ist, um eine bestimmte Menge an Wasserstoff und Sauerstoff zu erhalten Die Substanz hängt nicht von der Größe der Elektroden, dem Abstand zwischen ihnen und der Anzahl der Platten in der Batterie ab, die die Zelle versorgen. Darüber hinaus entdeckte Faraday, dass die Menge einer bei der Elektrolyse freigesetzten Substanz direkt proportional zu ihrem chemischen Äquivalent und der durch den Elektrolyten geleiteten Strommenge ist. (Ein chemisches Äquivalent ist die Grammzahl eines Elements oder einer Verbindung, die mit einem Mol Atom (1,0078 g) Wasserstoff in Verbindungen reagiert oder dieses ersetzt; cm. Äquivalente Masse). Diese beiden grundlegenden Bestimmungen werden Faradaysche Gesetze genannt. Zusammen mit seinem Freund W. Whewell, einem Spezialisten für klassische Philologie, entwickelte Faraday auch eine neue Terminologie in der Elektrochemie. Er nannte in eine Lösung eingetauchte Leiter Elektroden (früher hießen sie Pole); führte die Konzepte „Elektrolyse“ (chemische Veränderungen im Zusammenhang mit dem Stromdurchgang), „Elektrolyt“ (leitende Flüssigkeit in elektrochemischen Zellen), „Anode“ (Elektrode, an der die Oxidationsreaktion stattfindet) und „Kathode“ (Elektrode, an der die Oxidationsreaktion stattfindet) ein es kommt zu einer Reduktionsreaktion). Er nannte Ladungsträger in Flüssigkeiten Ionen (vom griechischen „Wanderer“, „Wanderer“), und die Ionen, die sich zur Anode (positive Elektrode) bewegten, wurden „Anionen“ und diejenigen, die sich zur Kathode bewegten, „Kationen“ genannt. Faradays Forschungen zur elektromagnetischen Induktion führten zur Entwicklung elektrischer Generatoren, die die Durchführung elektrochemischer Prozesse im industriellen Maßstab ermöglichten.
Faraday erklärte die Fähigkeit von Lösungen, elektrischen Strom durchzulassen, mit der Anwesenheit von Ionen in ihnen, aber er selbst und andere Wissenschaftler wie I. Hittorf und F. Kohlrausch glaubten, dass Ionen unter dem Einfluss von Strom entstehen. Im Jahr 1884 schlug S. Arrhenius vor, dass Ionen tatsächlich einfach dann gebildet werden, wenn Salz in Wasser gelöst wird. Die Arbeiten von S. Arrhenius, J. Van't Hoff und W. Ostwald waren ein wichtiger Meilenstein in der Entwicklung der Elektrolyttheorie und Ideen über die physikalisch-chemischen Eigenschaften von Lösungen und ihre Thermodynamik. Die Übereinstimmung zwischen Theorie und experimentellen Daten zur Ionenleitfähigkeit und zu Gleichgewichten in Lösung wurde vollständiger, nachdem P. Debye und E. Hückel 1923 elektrostatische Wechselwirkungen zwischen Ionen über große Entfernungen berücksichtigten.
Einen wichtigen Beitrag zur elektrochemischen Thermodynamik und insbesondere zur Aufklärung der Natur des elektrischen Potentials (Spannung) in einer elektrochemischen Zelle und des Gleichgewichts zwischen elektrischer, chemischer und thermischer Energie leisteten J. Gibbs und W. Nernst. Das elektrochemische Potenzial wird durch die chemische Energie der in der Zelle ablaufenden Prozesse bestimmt, hängt aber auch von deren Geschwindigkeit (Kinetik) ab. Die Modellierung kinetischer Prozesse an Elektroden wurde von Y. Tafel (1905), J. Butler (1924), M. Volmer (1930) und A. N. Frumkin (1930–1933) durchgeführt.
Elektrochemische Zellen.
Eine elektrochemische Zelle besteht normalerweise aus zwei Halbzellen, von denen jede eine in ihren eigenen Elektrolyten eingetauchte Elektrode darstellt. Elektroden bestehen aus elektrisch leitendem Material (Metall oder Kohlenstoff) oder seltener aus einem Halbleiter. Die Ladungsträger in den Elektroden sind Elektronen und die Ladungsträger im Elektrolyten sind Ionen. Eine wässrige Lösung von Kochsalz (Natriumchlorid NaCl), einem Elektrolyten, enthält geladene Teilchen: Natriumkationen Na + und Chloranionen Cl –. Wenn Sie eine solche Lösung in ein elektrisches Feld bringen, bewegen sich die Na + -Ionen zum negativen Pol und die Cl – -Ionen zum positiven Pol. Auch geschmolzene Salze wie NaCl sind Elektrolyte. Elektrolyte können beispielsweise auch Feststoffe sein B- Aluminiumoxid (Natriumpolyaluminat), das mobile Natriumionen oder Ionenaustauschpolymere enthält.
Die Halbzellen sind durch eine Trennwand getrennt, die die Bewegung der Ionen nicht behindert, aber eine Vermischung der Elektrolyte verhindert. Die Rolle einer solchen Trennwand kann eine Salzbrücke, ein an beiden Enden mit Glaswolle verschlossenes Rohr mit einer wässrigen Lösung, eine Ionenaustauschermembran oder eine poröse Glasplatte übernehmen. Beide Elektroden der Elektrolysezelle können in den gleichen Elektrolyten eingetaucht sein.
Es gibt zwei Arten von elektrochemischen Zellen: galvanische Zellen und elektrolytische Zellen (Elektrolyseure). In einer galvanischen Zelle finden chemische Reaktionen spontan an der Grenzfläche zwischen Elektrode und Elektrolyt statt, und die Elektroden sind durch einen Leiter miteinander verbunden. Mehrere in Reihe geschaltete galvanische Zellen bilden eine Batterie – eine chemische Stromquelle. In einer Elektrolysezelle finden aufgrund einer externen elektrischen Energiequelle Reaktionen an der Grenzfläche zwischen Elektrode und Elektrolyt statt. Letztere wird in chemische Energie der an den Elektroden entstehenden Reaktionsprodukte umgewandelt. Der Aufbau der galvanischen Zelle ist in Abb. dargestellt. 1 und der Elektrolyseur - in Abb. 2. Beachten Sie, dass sich dieselbe Zelle je nach Betriebsart entweder als galvanische Zelle oder als Elektrolyseur verhalten kann. Somit fungiert eine Blei-Säure-Autobatterie als galvanische Zelle, wenn sie zum Starten des Motors (während der Entladung) verwendet wird, und als Elektrolyseur, wenn sie über einen Autogenerator oder ein Ladegerät geladen wird.
Eine einfache galvanische Zelle, die 1836 von J. Daniel geschaffen wurde (Abb. 1), besteht aus zwei Elektroden: Zink, eingetaucht in eine wässrige Lösung von Zinksulfat, und Kupfer, eingetaucht in eine wässrige Lösung von Kupfer(II)sulfat. Ein solches Element ähnelt Kupfer-Zink-Paaren in einer Voltaiksäule. Beim Schließen des äußeren Stromkreises werden die Zinkatome auf der Oberfläche der Zinkelektrode unter Freisetzung von Elektronen zu Ionen oxidiert: Zn ® Zn 2+ + 2e – . Diese Elektronen bewegen sich entlang des äußeren Stromkreises zur Kupferelektrode und reduzieren Kupferionen zu Atomen: Cu 2+ + 2e – ® Cu. Der Elektronenfluss im externen Stromkreis ist der vom Element erzeugte Strom. Die Gesamtreaktion, die zu einer chemischen Umwandlung und der Erzeugung elektrischer Energie führt, hat die Form
Genau die gleiche Reaktion findet statt, wenn metallisches Zink zu einer Kupfersulfatlösung gegeben wird, allerdings wird in diesem Fall die chemische Energie in thermische Energie umgewandelt.
Elektrochemische Zellen werden oft schematisch dargestellt, indem die Grenze zwischen Elektrode und Elektrolyt mit einem Vertikalen oder Schrägstrich (| oder /) und die Salzbrücke mit zwei Schrägstrichen (//) gekennzeichnet wird. Die galvanische Zelle in Abb. 1 Antworteintrag
wobei M die molare Konzentration der Lösung ist.
In der in Abb. 2 laufen die gleichen Reaktionen ab wie in industriellen Elektrolyseuren zur Herstellung von Chlor und Alkali: die Umwandlung von Sole (einer konzentrierten wässrigen Lösung von Natriumchlorid) in Chlor und Natriumhydroxid NaOH:
Chloridionen an der Graphitelektrode werden zu Chlorgas oxidiert und Wasser an der Eisenelektrode wird zu Wasserstoff- und Hydroxidionen reduziert. Elektrolyte bleiben aufgrund der Bewegung von Natriumionen durch eine Trennwand – eine Ionenaustauschmembran – elektrisch neutral. Die Elektrode, an der die Oxidation stattfindet (Zink in Abb. 1 und Graphit in Abb. 2), wird Anode genannt, und die Elektrode, an der die Reduktion stattfindet, wird Kathode genannt.
Elektrodenpotential.
Die wichtigsten elektrischen Parameter elektrochemischer Zellen sind Strom (gemessen in Ampere, A) und Potenzial (gemessen in Volt, V). Die Stromstärke wird durch die Geschwindigkeit der Elektrodenreaktionen und das Potential durch die chemische Energie der in der Zelle ablaufenden Prozesse bestimmt. Sie ist gleich der Energie (gemessen in Joule, J) geteilt durch die Elektrizitätsmenge (gemessen in Coulomb, C), d. h. 1 V = 1 J / Cl. Daher ist das Potenzial eines Elements (elektromotorische Kraft, EMK) ein Maß für die Energie, die bei den in ihm ablaufenden Reaktionen erzeugt wird. Bei offenem Außenstromkreis treten keine Elektrodenreaktionen auf.
Das Potenzial einer galvanischen Zelle mit offenem Außenkreis gibt Aufschluss über die Thermodynamik ihrer Reaktionen. Das Potential des in Abb. gezeigten Elements. 1, bei Lösungskonzentrationen von 1 M und einer Temperatur von 25 °C - seinem Standardpotential E° – entspricht 1,10 V. Entsprechende Energie, thermodynamisches Gibbs-Potenzial, D G°, ist gegeben durch
Wo N– die Anzahl der während der Reaktion übertragenen Elektronen (in diesem Fall 2), F– Faradaysche Zahl (96 485 C / Mol). Das Potenzial einer galvanischen Zelle ist gleich der Potenzialdifferenz zwischen ihren beiden Halbzellen, d. h. die Differenz seiner Elektrodenpotentiale. Elektrodenpotentiale werden relativ zum Potential der Referenzelektrode gemessen, das herkömmlicherweise als Null angenommen wird ( cm. Tisch). Als Standardelektrode wurde nach Vereinbarung eine Normalwasserstoffelektrode (N.H.E.) gewählt; Es handelt sich um eine Platinplatte, die mit Platinschwarz beschichtet ist, mit Wasserstoffgas bei einem Druck von 1,01H 10 5 Pa (1 atm) gesättigt und in eine Lösung eingetaucht ist, die H + -Ionen mit thermodynamischer Aktivität enthält A= 1. Schematisch kann diese Elektrode als Pt/H 2 (1,01H 10 5 Pa)/H + ( A= 1). Darauf findet die Reaktion 2H + + 2e – ® H 2 statt. Um das Standardpotential einer Cu/Cu 2+ Kupferelektrode zu bestimmen, bauen Sie die folgende galvanische Zelle zusammen:
und für die Halbreaktion Cu 2+ + 2e – ® Cu ergibt die Messung
Ebenso erhalten wir für die Zn/Zn 2+ Halbzelle, in der die Reaktion Zn 2+ + 2e – ® Zn stattfindet
Die Differenz zwischen diesen beiden Standardelektrodenpotentialen entspricht dem Standardpotential des Zn-Cu-Elements.
Tatsächlich wird die Wasserstoffelektrode bei potentiometrischen Messungen selten verwendet, da sie ein ideales System darstellt, das in der Praxis nur schwer umzusetzen ist. Viel häufiger werden komfortablere und kompaktere Vergleichselektroden verschiedener Typen verwendet, die einen spezifischen, sorgfältig gemessenen Potentialwert im Verhältnis zum Strom aufweisen. Normalerweise verwenden sie eine Kalomelelektrode (CE), bestehend aus metallischem Quecksilber, Quecksilberchlorid (Kalomel) und einer Kaliumchloridlösung: Hg /Hg 2 Cl 2 /KCl. An der Elektrode findet folgende Reaktion statt:
Potenzial k.e. hängt von der Konzentration der Quecksilberionen ab, und diese hängt von der Konzentration der KCl-Lösung ab. Für gesättigte KCl-Lösung E° (k.e.) av = 0,2412 V (n.e.) bei 25° C.
|
STANDARDELEKTRODENPOTENZIALE |
|
| Elektrodenreaktion |
E o, V |
| Li + + e – ® Li | |
| Mg 2+ + 2e – ® Mg | |
| Al 3+ + 3e – ® Al | |
| Zn 2+ + 2e – ® Zn | |
| Cr 3+ + e – ® Cr 2+ | |
| 2H + + 2e – ® H 2 | |
| Cu 2+ + 2e – ® Cu | |
| Fe 3+ + e – ® Fe 2+ | |
| O 2 + 4H + + 4e – ® 2H 2 O | |
| Cl 2 + 2e – ® 2Cl – | |
| F 2 + 2e – ® 2F – | |
Beachten Sie, dass die Substanz einiger Elektroden nicht in die Gleichung der entsprechenden Reaktion einbezogen wird. Ja, Reaktion
tritt tatsächlich an einer Platinelektrode in einer Pt/Fe 3+, Fe 2+-Zelle auf. Die Platinelektrode ist inert und stellt nur Kontakt mit dem Elektrolyten her, der die oxidierten und reduzierten Formen des Elements (in diesem Fall zwei- und dreiwertige Eisenionen) enthält. Die Platinelektrode spielt bei N.V.E. die gleiche Rolle.
Mithilfe von Elektrodenpotentialtabellen können Sie die EMF einer galvanischen Zelle anhand ihrer Elektrodenpotentiale berechnen. Sie können auch verwendet werden, um vorherzusagen, ob eine bestimmte Redoxreaktion stattfinden wird. Von einem Standardelektrodenpotential kann nur dann gesprochen werden, wenn die Aktivität der an der Reaktion beteiligten Komponenten gleich 1 ist, d.h. ihre Konzentration in Lösung liegt nahe bei 1 M. Elektrodenpotential E hängt von der Konzentration der oxidierten und reduzierten Formen in Lösung ab und hängt mit diesen und dem Standardpotential zusammen E° Nernst-Gleichung. Für eine allgemeine Reaktion
Ochse + N e – = rot
Diese Gleichung hat die Form
Wo R- Universelle Gas Konstante, T– absolute Temperatur und – Aktivitäten oxidierter und reduzierter Formen. Die Aktivitäten reiner Feststoffe und Flüssigkeiten werden mit 1 angenommen. Bei 25° C RT/F= 0,025 V. Durch Messung der Elektrodenpotentiale relativ zum Potential der Referenzelektrode ist es möglich, die Konzentrationen von Stoffen in der Lösung anhand von Gleichung (10) zu bestimmen; Diese Methode wird Potentiometrie genannt.
Elektrodenreaktionen.
Potentiometrische Messungen werden unter Bedingungen durchgeführt, bei denen in der elektrochemischen Zelle kein Strom fließt. Dies bedeutet, dass darin keine allgemeinen chemischen Veränderungen auftreten und das gemessene Potential (Gleichgewicht) durch die Thermodynamik der Reaktionen bestimmt wird. Unter diesen Bedingungen haben Faktoren wie die Größe und Form der Elektroden oder die Intensität des Rührens der Lösung keinen Einfluss auf das gemessene Potential. Fließt Strom durch eine elektrochemische Zelle, so hängt die Geschwindigkeit der Elektrodenreaktionen nicht nur von thermodynamischen Parametern, sondern gemäß der Gleichung auch von der Stromstärke ab
Wo N– Anzahl der an einer bestimmten Elektrodenreaktion beteiligten Elektronen, F– Faradaysche Zahl. In diesem Fall hängt das Potenzial der elektrochemischen Zelle von kinetischen Faktoren sowie vom Material, aus dem die Elektrode besteht, der Größe und Form der Elektrode, der Intensität des Rührens der Lösung und vielen anderen Faktoren ab. Der Innenwiderstand der Zelle darf nicht vernachlässigt werden. Zusätzlich zur Potentialdifferenz an beiden Elektroden-/Elektrolytgrenzen tritt in der Lösung selbst aufgrund ihres Widerstands ein Spannungsabfall auf. Dieser Spannungsabfall erschwert die Untersuchung der Auswirkungen der an beiden Elektroden ablaufenden Reaktionen. Normalerweise wird die Reaktion an einer Elektrode, die als Arbeits- oder Indikatorelektrode bezeichnet wird, mithilfe einer Zelle mit drei Elektroden untersucht (Abb. 3): Die dritte Elektrode (z. B. gesättigtes Kalomel) wird im selben Fach wie die Arbeitselektrode platziert eins, so nah wie möglich daran, um den Effekt des ohmschen Spannungsabfalls zu minimieren. Durch die Messung des Stroms durch die Arbeitselektrode in Abhängigkeit vom Potential dieser Elektrode gegenüber der Referenzelektrode wird die sogenannte Polarisationskurve.
Wenn ein externer Strom fließt, weicht das Elektrodenpotential vom Gleichgewichtspotential ab. Diese Abweichung wird Polarisation genannt, ihr Betrag Überspannung. Überspannung hängt von mehreren Faktoren ab, die die Geschwindigkeit der Elektrodenreaktionen begrenzen. Schnelle Elektrodenreaktionen bei einer gegebenen Stromdichte (Stromstärke pro Einheit Elektrodenfläche) treten bei Potentialen nahe der Thermodynamik und daher bei geringer Überspannung auf. Langsame Reaktionen zeichnen sich durch hohe Überspannung aus. Die Geschwindigkeit der Elektrodenreaktionen und damit die Überspannung hängen von der Konzentration der Reagenzien, der Temperatur, dem Lösungsmittel, dem Elektrodenmaterial, der Methode und Geschwindigkeit des Stoffübergangs sowie der Stromdichte ab. Die Gesamtüberspannung lässt sich in mehrere Komponenten zerlegen: Konzentration, Aktivierung und Reaktion.
Eine Konzentrationsüberspannung entsteht dadurch, dass sich bei Stromdurchgang die Konzentration des reagierenden Ions auf der Oberfläche der Elektrode ändert, da in diesem Bereich elektroaktive Substanzen verbraucht werden und Reaktionsprodukte entstehen. Betrachten wir die Reduktion von Cu 2+ an einer Kupferelektrode. Die Konzentration an Cu 2+ in der Lösung beträgt zunächst 1 M. Wenn im Stromkreis kein Strom vorhanden ist, liegt das Potenzial der Kupferelektrode nahe am Standardpotenzial des Paares Cu/Cu 2+, d. h. 0,34 V relativ zu N.E. [ cm. Gleichung (6)]. Mit dem Durchgang des Kathodenstroms nimmt die Konzentration der Cu 2+ -Ionen auf der Elektrodenoberfläche ab und das Kathodenpotential wird gemäß der Nernst-Gleichung (10) zunehmend negativ. Frische Teile des Reagenzes gelangen auf unterschiedliche Weise von der Lösung zur Elektrode: durch Diffusion, Konvektion und Migration. Je höher die Geschwindigkeit dieser Prozesse ist (z. B. je intensiver die Durchmischung), desto geringer ist die Konzentrationsüberspannung. Dieser Anteil der Gesamtüberspannung lässt sich oft berechnen. Wenn die Konzentrationspolarisation den Hauptbeitrag zur Gesamtüberspannung leistet (das bedeutet, dass die Geschwindigkeit der verbleibenden Stufen der Elektrodenreaktion hoch ist), wird die Reaktion als reversibel oder Nernstsche Reaktion bezeichnet.
Eine Aktivierungsüberspannung entsteht dadurch, dass der Elektronentransfer auf der Elektrodenoberfläche nicht sofort, sondern mit endlicher Geschwindigkeit erfolgt. Betrachten wir die verallgemeinerte Elektrodenreaktion ox + N e – = rot. Um Elektronen mit einer bestimmten Geschwindigkeit (d. h. bei einer bestimmten Stromdichte) auf oxidierte Verbindungen zu übertragen, ist es notwendig, eine Energiebarriere zu überwinden, die als Aktivierungsenergie der Elektrodenreaktion bezeichnet wird. Diese Energie wird durch das angelegte Potenzial bereitgestellt. Der Zusammenhang zwischen Stromdichte und Aktivierungsüberspannung wird durch die Butler-Volmer- und Tafel-Gleichungen beschrieben, mit denen sich die kinetischen Parameter von Elektrodenreaktionen bestimmen lassen. Viele Elektrodenreaktionen, wie die Reduktion von Wasser zu Wasserstoff und seine Oxidation zu Sauerstoff, laufen langsam ab. Die Geschwindigkeit der Elektrodenreaktion kann stark vom Material, aus dem die Elektrode besteht, und den Eigenschaften ihrer Oberfläche abhängen. Beispielsweise ist an einer Quecksilberelektrode die Reduktion von Wasser zu Wasserstoff erheblich schwierig: Sie ist durch eine hohe Wasserstoffüberspannung gekennzeichnet. Diese Reaktion läuft auf Platin viel schneller und mit weniger Überspannung ab; Aus diesem Grund wird in der Wasserstoff-Referenzelektrode Platin verwendet. Die Reaktionsgeschwindigkeit hängt auch von den Substanzen ab, die an der Elektrodenoberfläche adsorbiert oder gebunden werden; Somit verringern Cyanidionen und eine Reihe organischer Verbindungen die Geschwindigkeit der Wasserstoffentwicklung auf der Oberfläche der Platinelektrode. Gleichzeitig können einige Tenside die Geschwindigkeit der Elektrodenreaktion deutlich erhöhen – sie werden Elektrokatalysatoren genannt.
Eine Reaktionsüberspannung tritt auf, wenn die Übertragung von Elektronen an der Elektrode mit einer chemischen Reaktion in der Lösung gekoppelt ist. Eine solche Reaktion kann als Quelle für am Elektronentransfer beteiligte Partikel dienen und gleichzeitig die Geschwindigkeit des gesamten Elektrodenprozesses begrenzen. Aus diesem Grund ist es so wichtig, die Details des Mechanismus (d. h. Stufen und Zwischenzustände) von Elektrodenreaktionen zu kennen. In vielen Fällen durchläuft das Ausgangsmaterial mehrere Umwandlungen, bevor es an der Elektrode zum Endprodukt wird, wie im Fall der Reduktion von Sauerstoff zu Wasser, einem Prozess von großer praktischer Bedeutung. Die Gesamtreaktion hat die Form
und besteht aus mehreren Stufen, in denen in einer Stufe die Sauerstoff-Sauerstoff-Bindung aufgebrochen wird. Aufgrund dieser mehrstufigen Reaktion verläuft die Reaktion an den meisten Elektroden langsam und wird im industriellen Maßstab in Gegenwart von Elektrokatalysatoren durchgeführt. Der Mechanismus von Elektrodenreaktionen wird mit den unten beschriebenen elektroanalytischen Methoden untersucht. Oft ändert sich der Verlauf einer Reaktion, wenn sich die Zusammensetzung der Lösung und die Art des Lösungsmittels ändern. Beispielsweise verläuft die Sauerstoffreduktion in Acetonitril bei Protonenmangel nach einem einfachen Ein-Elektronen-Mechanismus:
Elektrochemische Analysemethoden.
Für die qualitative und quantitative Analyse chemischer Substanzen wurden verschiedene elektrochemische Methoden entwickelt, die häufig auch zur Bestimmung der thermodynamischen und kinetischen Parameter von Elektrodenreaktionen und zur Untersuchung ihrer Mechanismen nützlich sind.
Konduktometrie
basiert auf der Messung der elektrischen Leitfähigkeit einer Lösung und dient zur Bestimmung der Konzentration von Salzen, Säuren, Basen usw. Bei konduktometrischen Bestimmungen werden üblicherweise Elektroden aus identischen Materialien verwendet und die Bedingungen für deren Leitung werden so gewählt, dass der Beitrag von Potentialsprüngen an beiden Elektroden-Elektrolyt-Grenzflächen minimiert wird (z. B. wird hochfrequenter Wechselstrom verwendet). ). Den Hauptbeitrag zum gemessenen Zellpotential leistet dabei der ohmsche Spannungsabfall IR, Wo R– Lösungsbeständigkeit. Die elektrische Leitfähigkeit einer Einkomponentenlösung kann mit ihrer Konzentration in Beziehung gesetzt werden, und die Messung der elektrischen Leitfähigkeit von Elektrolyten mit komplexer Zusammensetzung ermöglicht die Abschätzung des Gesamtionengehalts in der Lösung und wird beispielsweise zur Überwachung der Qualität von Destillaten verwendet oder entionisiertes Wasser. Bei einer anderen Art der Konduktometrie – der konduktometrischen Titration – wird der analysierten Lösung ein bekanntes Reagens portionsweise zugesetzt und die Änderung der elektrischen Leitfähigkeit überwacht. Der Äquivalenzpunkt, bei dem eine starke Änderung der elektrischen Leitfähigkeit festgestellt wird, wird aus einem Diagramm der Abhängigkeit dieses Werts vom Volumen des zugesetzten Reagenzes bestimmt.
Potentiometrie
Wird zur Bestimmung verschiedener physikalisch-chemischer Parameter auf der Grundlage von Daten zum Potenzial einer galvanischen Zelle verwendet. Das Elektrodenpotential bei fehlendem Strom im elektrochemischen Schaltkreis, gemessen relativ zur Referenzelektrode, steht durch die Nernst-Gleichung in Beziehung zur Konzentration der Lösung. Bei potentiometrischen Messungen werden häufig ionenselektive Elektroden verwendet, die hauptsächlich auf ein Ion in der Lösung empfindlich sind: eine Glaselektrode zur Messung des pH-Werts und Elektroden zur selektiven Bestimmung von Natrium-, Ammonium-, Fluor-, Calcium-, Magnesium- usw. Ionen. Die Oberfläche Die Schicht der ionenselektiven Elektrode kann Enzyme enthalten, und das Ergebnis ist ein System, das gegenüber dem entsprechenden Substrat empfindlich ist. Beachten Sie, dass das Potential einer ionenselektiven Elektrode nicht wie bei Stoffen mit elektronischer Leitfähigkeit durch die Übertragung von Elektronen bestimmt wird, sondern hauptsächlich durch die Übertragung oder den Austausch von Ionen. Die Nernst-Gleichung, die das Elektrodenpotential mit dem Logarithmus der Konzentration (oder Aktivität) einer Substanz in Lösung in Beziehung setzt, ist jedoch auch auf eine solche Elektrode anwendbar. Bei der potentiometrischen Titration wird das Reagens portionsweise der zu analysierenden Lösung zugesetzt und die Potentialänderung überwacht. Die für diese Art der Titration charakteristischen S-förmigen Kurven ermöglichen die Bestimmung des Äquivalenzpunkts und die Ermittlung thermodynamischer Parameter wie der Gleichgewichtskonstante und des Standardpotentials.
Voltammetrie.
Alle Arten voltammetrischer Methoden verwenden eine funktionierende Mikroelektrode mit einer Oberfläche von 10–7–10–1 cm2. Die damit gewonnenen Strom-Spannungs-Kurven ermöglichen die Identifizierung gelöster Stoffe, die Bestimmung ihrer Konzentration und häufig auch thermodynamischer und kinetischer Parameter. Die erste voltammetrische Methode – die Polarographie – wurde 1922 vom tschechischen Chemiker J. Heyrovsky vorgeschlagen. Die Arbeitselektrode in seinem Aufbau war eine tropfende Quecksilberelektrode. Quecksilber hat eine hohe Wasserstoffüberspannung, daher eignet sich eine Quecksilberelektrode zur Untersuchung von Prozessen, die bei negativen Potentialen ablaufen. Die Elektrodenoberfläche wird während des Messvorgangs ständig erneuert, wodurch eine Verschmutzung der Elektrode vermieden wird. Voltammetrische Untersuchungen werden auch mit Festkörperelektroden wie Platin und Kohlenstoff durchgeführt und nutzen Prozesse, die bei positiven Potentialen ablaufen. Bei der linearen Potentialdurchlaufvoltammetrie (Chronoamperometrie) ändert sich das Potential linear mit der Zeit und die Lösung wird nicht gerührt, sodass der Stofftransfer ausschließlich durch Diffusion erfolgt. Bei der Cyclovoltammetrie werden wiederholte dreieckige Spannungsimpulse an eine Elektrode angelegt. Die im aufsteigenden Abschnitt des Zyklus gebildeten Stoffe werden im absteigenden Abschnitt untersucht. Diese Methode ist besonders effektiv für die Untersuchung des Mechanismus von Elektrodenreaktionen durch die Analyse von Polarisationskurven bei unterschiedlichen Potund unterschiedlichen Lösungskonzentrationen. Es gibt andere Arten der Voltammetrie – Differenzimpuls und Rechteckwelle – bei denen Spannungsimpulse unterschiedlicher Form einem linear ansteigenden Potential überlagert werden. Diese Methoden werden häufig verwendet, um kleine Konzentrationen von Substanzen in Lösung zu bestimmen. Kommt es bei einer voltammetrischen Messung zu einer Durchmischung der Lösung, das heißt, dass ein Stoffaustausch gleichzeitig durch Konvektion und Diffusion erfolgt, spricht man von hydrodynamischer Voltammetrie. In diesem Fall ist es sinnvoll, eine rotierende Scheibenelektrode zu verwenden, da die experimentellen Strom-Spannungs-Kurven direkt mit den theoretischen verglichen werden können.
Amperometrie.
Die Methode basiert auf der Messung des Grenzdiffusionsstroms, der bei einer festen Spannung zwischen der Indikatorelektrode und der Referenzelektrode durch eine Lösung fließt. Bei der amperometrischen Titration wird der Äquivalenzpunkt durch den Bruch in der Stromkurve bestimmt – das Volumen der zugegebenen Arbeitslösung. Chronoamperometrische Methoden basieren auf der Messung der Stromabhängigkeit von der Zeit und werden hauptsächlich zur Bestimmung von Diffusionskoeffizienten und Geschwindigkeitskonstanten eingesetzt. Elektrochemische Miniaturzellen, die als Sensoren am Ausgang von Flüssigchromatographensäulen dienen, arbeiten nach dem Prinzip der Amperometrie (sowie der Voltammetrie). Galvanostatische Methoden ähneln amperometrischen Methoden, messen jedoch das Potenzial, wenn eine bestimmte Strommenge durch die Zelle fließt. Somit wird bei der Chronopotentiometrie die zeitliche Änderung des Potenzials kontrolliert. Diese Methoden werden hauptsächlich zur Untersuchung der Kinetik von Elektrodenreaktionen verwendet.
Coulometrie.
Bei der Coulometrie erfolgt bei kontrolliertem Potential die vollständige Elektrolyse einer Lösung durch intensives Mischen in einem Elektrolyseur mit einer relativ großen Arbeitselektrode (unteres Quecksilber- oder Platinnetz). Gesamtstrommenge ( Q, C) für die Elektrolyse erforderlich ist, hängt von der Menge der sich bildenden Substanz ab ( A, d) Faradaysches Gesetz:
Wo M- Sie sagen Masse (g/mol), F- Faraday-Zahl. Bei der coulometrischen Titration wird mithilfe eines konstanten Stroms elektrolytisch ein Reagens erzeugt, das mit der zu bestimmenden Substanz reagiert. Der Verlauf der Titration wird potentiometrisch oder amperometrisch gesteuert. Coulometrische Methoden sind praktisch, weil sie absoluter Natur sind (d. h. sie ermöglichen die Berechnung der Analytmenge, ohne auf Kalibrierungskurven zurückgreifen zu müssen) und unempfindlich gegenüber Änderungen der Elektrolysebedingungen und Elektrolyseurparameter (Elektrodenoberfläche oder Rührintensität) sind. Bei der Coulogravimetrie wird die Menge der elektrolysierten Substanz durch Wiegen der Elektrode vor und nach der Elektrolyse bestimmt.
Es gibt andere elektroanalytische Methoden. Bei der Wechselstrompolarographie wird eine sinusförmige Spannung niedriger Amplitude an ein über einen weiten Frequenzbereich linear variierendes Potential angelegt und entweder die Amplitude und Phasenverschiebung des resultierenden Wechselstroms oder die Impedanz bestimmt. Aus diesen Daten werden Informationen über die Natur der gelösten Stoffe sowie über den Mechanismus und die Kinetik von Elektrodenreaktionen gewonnen. Bei Dünnschichtverfahren werden elektrochemische Zellen mit einer Elektrolytschicht von 10–100 µm Dicke eingesetzt. In solchen Zellen läuft die Elektrolyse schneller ab als in herkömmlichen Elektrolyseuren. Zur Untersuchung von Elektrodenprozessen werden spektrochemische Methoden mit spektrophotometrischer Registrierung eingesetzt. Zur Analyse von Substanzen, die sich auf der Oberfläche der Elektrode bilden, wird deren Lichtabsorption im sichtbaren, UV- und IR-Bereich gemessen. Veränderungen der Eigenschaften der Elektrodenoberfläche und des Mediums werden mit Methoden der elektrischen Reflexion und Ellipsometrie überwacht, die auf der Messung der Reflexion der Strahlung von der Elektrodenoberfläche basieren. Dazu gehören Methoden der Spiegelreflexion und Raman-Streuung von Licht (Raman-Spektroskopie) sowie der Spektroskopie der zweiten Harmonischen (Fourier-Spektroskopie).
Andere elektrochemische Phänomene und Methoden.
Bei der Relativbewegung von Elektrolyt und geladenen Teilchen oder Oberflächen treten elektrokinetische Effekte auf. Ein wichtiges Beispiel dieser Art ist die Elektrophorese, bei der die Trennung geladener Teilchen (z. B. Proteinmoleküle oder kolloidale Teilchen) erfolgt, die sich in einem elektrischen Feld bewegen. Elektrophoretische Methoden werden häufig zur Trennung von Proteinen oder Desoxyribonukleinsäuren (DNA) in Gelen eingesetzt. Elektrische Phänomene spielen eine große Rolle im Funktionieren lebender Organismen: Sie sind für die Erzeugung und Ausbreitung von Nervenimpulsen, das Auftreten von Transmembranpotentialen usw. verantwortlich. Zur Untersuchung biologischer Systeme und ihrer Bestandteile werden verschiedene elektrochemische Methoden eingesetzt. Es ist auch von Interesse, die Wirkung von Licht auf elektrochemische Prozesse zu untersuchen. Gegenstand der photoelektrochemischen Forschung ist daher die Erzeugung elektrischer Energie und die Auslösung chemischer Reaktionen unter Lichteinfluss, was für die Effizienzsteigerung bei der Umwandlung von Sonnenenergie in elektrische Energie von großer Bedeutung ist. Üblicherweise werden hier Halbleiterelektroden aus Titandioxid, Cadmiumsulfid, Galliumarsenid und Silizium verwendet. Ein weiteres interessantes Phänomen ist die Elektrochemilumineszenz, d.h. Erzeugung von Licht in einer elektrochemischen Zelle. Es wird beobachtet, wenn sich an den Elektroden energiereiche Produkte bilden. Häufig wird der Prozess zyklisch durchgeführt, um sowohl oxidierte als auch reduzierte Formen einer bestimmten Verbindung zu erhalten. Ihre Wechselwirkung untereinander führt zur Bildung angeregter Moleküle, die unter Lichtemission in den Grundzustand übergehen.
Angewandte Elektrochemie.
Elektrochemie hat viele praktische Anwendungen. Mit Hilfe primärer galvanischer Zellen (Einwegelemente), die mit Batterien verbunden sind, wird chemische Energie in elektrische Energie umgewandelt. Sekundärstromquellen – Batterien – speichern elektrische Energie. Brennstoffzellen sind primäre Energiequellen, die durch die kontinuierliche Zufuhr von Reaktanten (wie Wasserstoff und Sauerstoff) Strom erzeugen. Diese Prinzipien liegen tragbaren Stromquellen und Batterien zugrunde, die in Raumstationen, Elektrofahrzeugen und elektronischen Geräten verwendet werden.
Die großtechnische Produktion vieler Stoffe basiert auf der elektrochemischen Synthese. Durch die Elektrolyse von Sole im Chlor-Alkali-Verfahren entstehen Chlor und Alkali, die dann zur Herstellung organischer Verbindungen und Polymere sowie in der Zellstoff- und Papierindustrie verwendet werden. Die Produkte der Elektrolyse sind Verbindungen wie Natriumchlorat, Persulfat, Natriumpermanganat; Durch Elektroextraktion werden industriell wichtige Metalle gewonnen: Aluminium, Magnesium, Lithium, Natrium und Titan. Es ist besser, geschmolzene Salze als Elektrolyte zu verwenden, da in diesem Fall im Gegensatz zu wässrigen Lösungen die Reduktion von Metallen nicht durch die Freisetzung von Wasserstoff erschwert wird. Fluor wird durch Elektrolyse in geschmolzenem Salz hergestellt. Elektrochemische Prozesse dienen als Grundlage für die Synthese einiger organischer Verbindungen; Beispielsweise wird Adiponitril (ein Zwischenprodukt bei der Nylonsynthese) durch Hydrodimerisierung von Acrylnitril erhalten.
Das Galvanisieren von Silber, Gold, Chrom, Messing, Bronze und anderen Metallen und Legierungen wird häufig an verschiedenen Objekten praktiziert, um Stahlprodukte vor Korrosion zu schützen, zu dekorativen Zwecken, für die Herstellung von elektrischen Steckverbindern und Leiterplatten in der Elektronikindustrie. Elektrochemische Verfahren werden zur hochpräzisen Maßbearbeitung von Werkstücken aus Metallen und Legierungen eingesetzt, insbesondere solchen, die mit herkömmlichen mechanischen Verfahren nicht bearbeitet werden können, sowie zur Herstellung von Teilen mit komplexen Profilen. Wenn die Oberfläche von Metallen wie Aluminium und Titan eloxiert wird, bilden sich schützende Oxidfilme. Solche Filme entstehen auf der Oberfläche von Werkstücken aus Aluminium, Tantal und Niob bei der Herstellung von Elektrolytkondensatoren, manchmal auch zu dekorativen Zwecken.
Zur Untersuchung von Korrosionsprozessen und der Auswahl von Materialien, die diese Prozesse verlangsamen, werden häufig elektrochemische Methoden eingesetzt. Korrosion von Metallstrukturen kann durch kathodischen Schutz verhindert werden, bei dem eine externe Quelle an die zu schützende Struktur angeschlossen wird und die Anode und die Struktur auf einem Potential gehalten werden, das ihre Oxidation ausschließt. Die Möglichkeiten der praktischen Anwendung anderer elektrochemischer Verfahren werden untersucht. So kann Elektrolyse zur Reinigung von Wasser eingesetzt werden. Eine vielversprechende Richtung ist die Umwandlung von Sonnenenergie mittels photochemischer Methoden. Es werden elektrochemische Monitore entwickelt, deren Funktionsprinzip auf Elektrochemilumineszenz basiert. Erwähnenswert ist auch die Untersuchung reversibler Farbveränderungen der Elektrodenoberfläche als Folge von Elektrodenreaktionen.
Literatur:
Agladze R.I. Angewandte Elektrochemie. M. – L., 1975
Izmailov N.A. Elektrochemie von Lösungen. M., 1976
Messmethoden in der Elektrochemie, Bd. 1–2. M., 1977
Koryta I. Ionen, Elektroden, Membranen. M., 1983
Bagotsky V.S. Grundlagen der Elektrochemie. M., 1988